
Abstract
Background
Oral squamous cell carcinoma (OSCC) is one of the malignant tumors with a poor prognosis. Periodontitis (PD is considered a high-risk factor for OSCC, but the genetic mechanism is rarely studied. This study aims to link OSCC and PD by identifying common differentially expressed miRNAs (Co-DEmiRNAs), their related genes (Hub genes), transcription factors (TFs), signaling pathways, enrichment functions, and compounds, and searching for genetic commonalities.
Methods
The miRNAs expression datasets of OSCC and PD were searched from the GEO database. The miRNA and related crosstalk mechanism between OSCC and PD was obtained through a series of analyses.
Results
hsa-mir-497, hsa-mir-224, hsa-mir-210, hsa-mir-29c, hsa-mir-486-5p, and hsa-mir-31are the top miRNA nodes in Co-DEmiRNA-Target networks. The most significant candidate miRNA dysregulation genes are ZNF460, FBN1, CDK6, BTG2, and CBX6, while the most important dysregulation TF includes HIF1A, TP53, E2F1, MYCN, and JUN. 5-fluorouracil, Ginsenoside, Rh2, and Formaldehyde are the most correlated compounds. Enrichment analysis revealed cancer-related pathways and so on.
Conclusions
The comprehensive analysis reveals the interacting genetic and molecular mechanism between OSCC and PD, linking both and providing a foundation for future basic and clinical research.
Similar content being viewed by others


Introduction
Oral squamous cell carcinoma (OSCC) is one of the most common head and neck malignant tumors, with a high incidence of 350,000 new cases and 170,000 deaths [1]. The Asian region has the highest OSCC morbidity and mortality among all other countries, which various etiological factors may cause. The risk factors of OSCC recognized by researchers include smoking [2], drinking [3], chewing betel nut [4], periodontal disease [5], gene mutation [6], and so on. However, periodontal diseases represented by periodontitis (PD) have played an increasingly significant role in the occurrence and development of OSCC [7, 8].
There is a convincing correlation between inflammation and the occurrence and development of many cancers [9,10,11]. The role of inflammation in tumors can also be observed in the oral environment [12]. PD also is strongly associated with a variety of tumors, including breast cancer [13], pancreatic cancer [14], gastric cancer [15], and colorectal cancer [16]. To date, PD is considered one of the most common inflammatory conditions affecting the oral cavity and one of the risk factors for OSCC [17]. Systematic reviews have confirmed the previous association between OSCC and PD [18], but further mechanism study of the shared connotation still needs to be completed. In particular, little is known about the possible epigenetic mechanisms of OSCC and PD.
MicroRNA (miRNAs) are small RNAs that play vital roles in regulating gene expression. miRNAs regulate cellular physiological processes by regulating expression and play a crucial role in mediating diseases [19]. The molecular mechanism of disease occurrence and development can be further grasped by studying disease-related miRNAs and their expression patterns. In addition, miRNAs can serve as biomarkers of two diseases (OSCC and PD) or key targets for therapeutic drugs [20].
This study aimed to comprehensively analyze differentially expressed miRNAs (DEmiRNAs) in OSCC and PD to identify candidate CO-DEmiRNAs, their associated hub genes, signaling pathways and related compounds. Thereby promoting the understanding of the shared molecular mechanisms between closely related tumors and non-neoplastic diseases. And providing a theoretical basis for driving future basic research and clinical practice.
In more than 20 years of clinical work by our group, we found that almost all patients with OSCC suffer from periodontitis while burdening the tumor. Based on our previous translational studies on inflammation-precancerous lesion cancer, we sought to explore whether the presence of crucial genetic molecules could serve as a connecting key for PD and OSCC. Recent studies have revealed numerous noncoding RNAs (ncRNAs) roles in cancer and various diseases, highlighting the biological significance of these previously “neglected” RNA species. In particular, microRNAs (miRNAs) are involved in many biological processes that affect cell homeostasis. MiRNAs are considered post-transcriptional gene regulators that can achieve translational repression, mRNA degradation, and gene silencing and play a significant role in gene expression. We sought to explore and determine whether there are co-expressed key miRNAs and transcription factors present in PD and OSCC by bioinformatics methods, thus providing a solid basis for our subsequent target findings. This helps us in a series of studies in stomatitis-cancer transformation. We promote an understanding of the shared molecular mechanisms between closely related tumors and non-neoplastic diseases. We provide a theoretical basis for future basic research and clinical practice.
Materials and methods
MicroRNA datasets selection and preparation
Download the miRNA expression datasets of OSCC and PD from the GEO database (Table 1) (https://www.ncbi.nlm.nih.gov/geo/). Only one OSCC miRNA dataset GSE45238 was identified for analysis (obtained from platform GPL8179, Illumina Human v2 MicroRNA expression bead chip). For the miRNA expression dataset for PD, we chose the most numerous GSE54710 (obtained from platform GPL15159, Agilent 031181 Unrestricted Human miRNA V16.0 Microarray 030840). All experiments were performed further with relevant guidelines and regulations.
Data processing and differential expression miRNA analysis
The microarray and expression data were downloaded using the R package “GEOquery” (https://www.r-project.org/). The data were corrected using the “ComBat” method in the R package “SVA.” The R package “Limma” was then used to identify miRNAs significantly differentially expressed in OSCC and PD cases and controls. miRNA (P value < 0.05 and |LogFC|> 0.5) are regarded as “DEmiRNA” and used for analysis. Furthermore, LogFC > 0.5 is overexpressed and LogFC < 0.5 is low expressed.
Shared DEmiRNA analysis and Co-DEmiRNA identification
The miRNA lists of the two diseases were processed using the R package “Venndiagram” to obtain Shared DEmiRNA. These were considered mutual DEmiRNAs and were further analyzed. We defined miRNAs with the common expression trend (both high/low expression) as Co-DEmiRNAs and excluded Shared DEmiRNAs with different expression trends (their opposite expression does not assist in disease-related studies, nor is it meaningful for scientific research).
Co-DEmiRNA-gene network construction and functional enrichment analysis
The co-DEmiRNA target network was constructed using miRNet 2.0 (https://www.mirnet.ca/). For the Co-DEmiRNA-Gene network, target genes were selected from 3 packages (TarBase v8.0), miRTarBase v8.0 (http://mirtarbase.mbc.nctu.edu.tw/php/index.php), and miRecords (http://c1.accurascience.com/miRecords). Due to poor stability, “Steiner Forest Network” cannot achieve the most stable link on the premise of ensuring the correlation. Instead, “Minimum Network” was chosen, which reduces the network complexity and retains key features that demonstrate network connectivity. It is computed using the critical nodes of all elements. To build the “Minimum Network”, the shortest paths between the nodes are determined, and any nodes not on the shortest path are removed.
Hub gene identification and functional enrichment analysis
From the constructed Co-DEmiRNA-Gene network, we selected key gene nodes with higher degrees and betweenness as “Hub Genes” that connect other parts of this complex network. In a sense, hub genes are the actual central complex members. In addition, we performed an enrichment analysis of hub genes for KEGG [21] and GO terms. Use the R package “ggplot2” for visualization and R package “clusterProfiler” to analyze selected data. The calculated P values were subjected to FDR correction for KEGG and GO enrichment, using FDR ≤ 0.05 as a threshold.
Co-DEmiRNA-TF network construction and functional enrichment analysis
We used multiple databases such as miRbase (https://www.mirbase.org/), TransmiR v2.0 (http://www.cuilab.cn/transmir) to download the corresponding transcription factors (TF) of Co-DEmiRNAs, extracted the corresponding TFs, and constructed a Co-DEmiRNA-TF Network using a similar method. The Co-DEmiRNA-TF networks were further subjected to functional enrichment analysis using the KEGG pathway, Reactome pathway, and GO.
Co-DEmiRNA-compound network construction
Search for small-molecule compounds with a high correlation with Co-DEmiRNAs. Build the Co-DEmiRNA-compound network based on data from CTD datasets (Comparative Toxicogenomic Database, http://ctdbase.org/), miRbase (https://www.mirbase.org/), SM2miR (http://bioinfo.hrbmu.edu.cn/SM2miR/) and PharmacomIR.
Co-DEmiRNA diagnostic efficacy analysis
Other databases were used to verify the diagnostic efficacy of the best Co-DEmiRNA as a diagnostic marker. The miRNA-seq data of the level 3 BCGSC miRNA Profiling in the TCGA (https://portal.gdc.cancer.gov/) HNSC (Head and Neck Squamous Cell Carcinoma) project were selected for verification, and the corresponding data without clinical information were discarded. Samples belonging to oral cancer sites (Alveolar Ridge, Tongue, Buccal Mucosa, Floor of mouth, Hard Palate, Oral Cavity) were retained in clinical information, and samples from nonoral cancer sites (Hypopharynx, Larynx, Lip, Oropharynx, Tonsil) were excluded. The miRNA-seq data in RPM (Reads per Million mapped reads) format was converted to log2, and 373 samples were obtained (using the R package “pROC” for data analysis and the “ggplot2” package for visualization). To calculate the area under the curve, the area value under the ROC curve should be between 0.5 and 1. The closer the AUC is to 1, the better the diagnostic effect.
The corresponding expression patterns of the two disease miRNAs are shown (Fig. 3). There were 18 shared DEmiRNAs with a similar expression trend (Table 2). 6 DEmiRNAs were co-overexpressed, while co-low expression was observed in 5 shared DEmiRNAs. The remaining seven shared DEmiRNAs showed diametrically opposite expression trends in the two diseases.
Results
DEmiRNA and shared DEmiRNA identification
In the OSCC dataset (GSE45238), the comparison was between the cases of OSCC (tumor specimens of OSCC patients) and normal cases (adjacent nontumor epithelium). After correction, 858 miRNAs were retained, and 208 OSCC-related significant DEmiRNAs were identified. In periodontitis data (GSE54710), the comparison was the case of periodontitis (periodontal tissue with periodontitis) and standard samples (healthy periodontal tissues). In contrast, PD dataset analysis retained 1368 miRNAs and identified 54 significant PD-related DEmiRNAs (Additional file 1: Table S1. a, b).
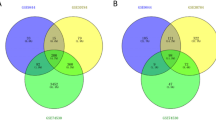
Compared with controls in OSCC-affected tissues, 103 DEmiRNAs were overexpressed, and 105 DEmiRNAs were under-expressed (Fig. 1A). Similarly, 35 miRNAs were overexpressed in PD-affected tissues, while 19 were under-expressed (Fig. 1B). A Venn diagram searched for the common part between the two DEmiRNA lists, and 18 shared DEmiRNAs were screened (Fig. 2). We also think the direct comparison of OSCC and periodontitis is important, and this could broaden our search for common molecular genetic mechanisms for both diseases in further research.

Data processing and differential expression analysis. A, B The Volcano map of OSCC and PD differentially expressed miRNA. The miRNA expression in OSCC is relatively broad and scattered, while PD is more concentrated (Blue indicates significant down-regulation, red indicates significant up-regulation, and black parts are non-statistically significant expressed genes.)

Shared DEmiRNA analysis. The intersection between the two graphs represents that there are 18 shared DEmiRNAs between OSCC (GSE45238) and PD (GSE54710)
The corresponding expression patterns of the two disease miRNAs are shown (Fig. 3). There were 18 shared DEmiRNAs with a similar expression trend (Table 2). 6 DEmiRNAs were co-overexpressed, while common low expression was observed in 5 shared DEmiRNAs. The remaining seven shared DEmiRNAs showed diametrically opposite expression trends in the two diseases.

The different expression trends of DEmiRNAs in OSCC (A) and PD (B). Only six shared miRNAs showed overexpression in both diseases, and 5 showed joint underexpression. (Red represents overexpression, and blue represents low expression. When the shared miRNA has the same color, it represents a common expression trend that it is has a common expression trend in OSCC and PD)
Co-DEmiRNA identification, Co-DEmiRNA-gene network and functional analysis
We selected 11 DEmiRNAs with the same expression trend as Co-DEmiRNAs for further analysis. They then constructed the Co-DEmiRNA-Gene Network. The minimum network consists of 63 genes and 22 miRNAs with 303 edges (Fig. 4, Additional file 1: Table S2a). The highest degree DEmiRNA nodes in the network are hsa-mir-497-5p, hsa-mir-224-5p, hsa-mir-210-3p, hsa-mir-29c-3p, hsa-mir-486-5p. The top 5 gene nodes with the highest degree in the network include ZNF460, FBN1, CDK6, BTG2, and CBX6. The most abundant signaling pathways are shown (Table 3. Additional file 1: Table S2b–f). KEGG pathway analysis showed Focal adhesion, ECM-receptor interaction Pathways in cancer, p53 signaling pathway, and other related pathways. Reactome analysis showed Signaling by SCF-KIT, Oncogene Induced Senescence, Pre-NOTCH Transcription Translation, PI3K/AKT pathway, and other related pathways. GO biology process (GO-BP) analysis showed negative regulation of the cellular process, Ras protein signal transduction, and so on. The most abundant GO molecular functions (GO-MF) include extracellular matrix structural constituents, transcription from RNA polymerase II promoters, and other binding-related functions, including growth factor, nucleotides, purine ribonucleotide, and purine nucleotides. The enriched top GO cellular components (GO-CC) include ruffle, nucleoplasm, cell leading edge, organelle, lumen, extracellular matrix, and other parts.

The minimum network of Co-DEmiRNA-Gene. The optimized and minimal network was used to obtain the five key miRNA nodes with the strongest centrality and the highest degree among the 11 DEmiRNAs, including hsa-mir-497-5p, hsa-mir-224-5p, hsa-mir-210-3p, hsa-mir-29c-3p, and hsa-mir-486-5p. The five most critical related gene nodes were also obtained, which were ZNF460, FBN1, CDK6, BTG2, and CBX6 (blue node: DEmiRNA, green node: gene)
Hub genes identification and enrichment function analysis
We obtained all the network's key node genes with statistical significance, including ZNF460, FBN1, CDK6, BTG2, CBX6, DYRK1A, and so on. We obtained some significant pathways corresponding to hub genes (Fig. 5). The figure shows that the PI3K/AKT signaling pathway was the most enriched KEGG pathway. In addition, GO-BP analysis reveals that Ras protein signal transduction and extracellular structure organization are the most enriched biological process. GO-CC analysis revealed that the enriched cellular components were collagen-containing extracellular matrix, cell leading edge, ruffle, and other parts. GO-MF analysis revealed that protein serine/threonine kinase activity, extracellular matrix structural constituents, and growth factor binding were the top enriched molecular functions.

KEGG pathway and GO enrichment analysis of Hub genes related to Co-DEmiRNAs. The stronger the correlation between miRNAs and pathways, the larger the number of counts and the larger the bubbles (The p value is determined by color. The closer the color is to red, the smaller the P value. P < 0.05 considered statistically significant)
Co-DEmiRNA-TF network and functional analysis
The corresponding transcription factors were queried by miRbase, and the key transcription factors of Co-DEmiRNAs were obtained (Additional file 1: Table S3). The original network consists of 48 TFs, eight miRNAs, and 64 edges. The minimum network consists of 9 TFs, eight miRNAs, and 21 edges (Fig. 6, Additional file 1: Table S4a). The top 5 transcription factors include HIF1A, TP53, E2F1, MYCN, and JUN, while hsa-mir-224 and hsa-mir-210 are the topmost miRNA nodes. The most abundant KEGG, Reactome, and GO pathways are listed (Table 4, Additional file 1: Table S4b–f). These include acute myeloid leukemia, pathways in cancer, and other pathways. GO-BP analysis revealed positive regulation of transcription from RNA polymerase II promoter and DNA-dependent. GO-MF analysis in TFs includes multiple transcription-related and binding-related functions. GO-CC analysis reveals that the most enriched cellular components are transcription factors in the complex, nucleoplasm, and other parts.

The minimum network of Co-DEmiRNA-TF. IF1A, TP53, E2F1, MYCN, and JUN are located close to the center, demonstrating that these 5 TFs are credible and central (blue node: Co-DEmiRNA, red node: TF)
Co-DEmiRNA-compound network and functional analysis
The query obtained several disease-related small molecule compounds (5-fluorouracil, Enoxacin 2, Cisplatin, etc.). A Co-DEmiRNA-Compound minimum network was constructed, consisting of 9 compounds and 11 miRNAs with 23 edges (Fig. 7, Additional file 1: Table S5). The most relevant miRNAs are hsa-mir-31, has-mir-224, has-mir-210, and has-mir-497. The nine compounds in this network include Formaldehyde, Ginsenoside Rh2, Trichostatin A (TSA), and Vincristine. The compound with the highest degree and betweenness is 5-fluorouracil, followed by Ginsenoside Rh2, Formaldehyde.

The minimum network of Co-DEmiRNA-Compound. In the optimal network, 5-fluorouracil, Arsenic trioxide, Cisplatin, Diethylstilbestrol, and Enoxacin were the compounds most strongly associated with known Co-DEmiRNAs (block: miRNA, circle: small molecule compound. The darker the color and larger the size of the key node in the figure, the higher its degree)
Key Co-DEmiRNAs/targets identification and diagnostic efficacy
The key Co-DEmiRNAs and targets were sorted out in Table 5. We selected the data of OSCC in TGCA as a validation set. The diagnostic efficacy of the top Hub Co-DEmiRNAs in OSCC was examined. The receiver operating characteristic (ROC) curves of these Co-DEmiRNA are depicted (Fig. 8). The predictive ability of variable hsa-mir-29c (AUC = 0.931) showed high accuracy, and the predictive ability of variables hsa-mir-497 (AUC = 0.819) and hsa-mir-486-5p (AUC = 0.7548) showed certain accuracy. The predictive ability of variable hsa-mir-224-5p (AUC = 0.912) showed high accuracy, hsa-mir-31-5p (AUC = 0.871) and variable hsa-mir-210-3p (AUC = 0.896) showed certain accuracy.

The ROC curve of top Co-DEmiRNAs. Co-DEmiRNAs, both co-up and co-down expressed, had a strong predictive ability for disease diagnosis in OSCC (AUC > 0.7, with larger values demonstrating that this miRNA has a strong predictive ability for the diagnosis of OSCC)
Discussion
The miRNA is an essential intermediate hub of host physiological and pathophysiological activities [33]. We know that the microbiota changes host miRNA using self-virulence factors, reducing the host immune response-ability, and achieving the final effect of pathogenicity [34,35,36]. Oral pathogens are important risk factors for periodontitis (PD) and oral squamous cell carcinoma (OSCC) [18, 37]. In recent years, it has been gradually discovered that PD-related pathogenic microorganisms, mainly Porphyromonas gingivalis (P. gingivalis) and Fusobacterium nucleatum (F. nucleatum), have played an essential role in oral cancer occurrence [38], which revealed to us that PD might also be the cause of OSCC or a key step in the malignant transformation process of oral disease. Overall, there may have a homologous genetic and molecular link between OSCC and PD.
Our current study explored the epigenetic mechanism of CO-DEmiRNA mediated the association between OSCC and PD by screening and identifying Co-DEmiRNA common in the two diseases. The network architecture was applied to determine DEmiRNA-related hub genes and TF, which could be used as the linkage mechanism of differential expression and further function of DEmiRNA. In addition, functional enrichment analysis was conducted on them to determine key pathways, molecular functions, and cell components. In addition, the small molecule compounds associated with Co-DEmiRNA were analyzed, and the key junction compounds between OSCC and PD were explored. The key Co-DEmiRNAs identified in this study may provide more effective guidance in the future study of inflammation-cancer transformation.
Most DEmiRNAs had the same expression trend in the two diseases, which further revealed the similar immune mechanism of the host oral microenvironment against inflammation or cancer, perhaps a common pattern of miRNA dysregulation in pro-inflammatory and pro-cancer responses. Co-DEmiRNAs with the highest degree included hsa-mir-224, hsa-mir-210, hsa-mir-31(overexpressed), and hsa-mir-497, hsa-mir-29c, hsa-mir-486(which were low expressed). They are all broadly involved in inflammation, cancer, and host immune responses.hsa-mir-224 is considered an early diagnostic marker of cancer [22], and both it and hsa-mir-210 are significantly involved in cancer progression and metastasis [23]. hsa-mir-31 is an important protective factor of the epithelial barrier [24] and has also been recognized as a cancer biomarker [25, 26]. hsa-mir-497 and has-mir-29c suppress various cancers, inhibiting the proliferation and growth of cancer [27,28,29,30]. hsa-mir-486 is a migration suppressor of various tumors and plays an important role in regulating epithelial-mesenchymal transition (EMT) [31, 32].
Our study revealed that dysregulation of associated gene expression mediated by noncoding RNA represented by miRNA might be the key mechanism linking PD to OSCC or other cancers. The genes with the highest degree in the Co-DEmiRNA-Gene network include ZNF460, FBN1, CDK6, BTG2, and CBX6, which may be the essential hub genes/mediators between OSCC and PD. ZNF460 (zinc finger protein 460) is involved in the regulation of multiple cancer processes by JAK2/STAT3 pathway [39], and its high expression is associated with the proliferation, invasion, and metastasis of colorectal cancer and oral cancer [39, 40]. FBN1 (fibrinin-1) is a common extracellular matrix encoding gene [41], and inactivation will affect the integrity of tissues (aortic wall, periodontal membrane, oral epithelial barrier, etc.). It encodes the formation of Oxytalan fibers [42], a unique component of the periodontal ligament (PDL). Low expression of FBN1 inhibits TGF-β 1-mediated expression of Periosteum, thereby inhibiting collagen fiber production. In addition, FBN1 also plays an important role in the Wnt/β-catenin signaling pathway that regulates cancer cell migration [43]. CDK6 (cyclin-dependent kinase 6), as one of the proto-oncogenes driving tumors, has become a key target of various cancer therapies [44], and its inhibition can significantly affect tumor cell metabolism and antitumor immunity [45, 46]. CDK6 also inhibits the proliferation of periodontal ligament cells (PDLCs) by regulating the cell cycle in periodontitis [47]. BTG2 (B cell translocation gene 2) has long been recognized as a tumor suppressor gene in various cellular processes [48,49,50], including cell division, DNA repair, transcriptional regulation, and messenger RNA stability. Upregulation of BTG2 inhibits cancer migration, invasion, EMT and, glycolysis [51]. CBX6 (chromobox protein 6) accelerates EMT in head and neck squamous cell carcinoma [52], resulting in cancer progression.
In the Co-DEmiRNA-TF network, transcription factors HIF1A, TP53, E2F1, MYCN, and JUN have the highest degree. HIF1A (hypoxia-induced transcription factor 1α) can promote gingival tissue aging and hypoxia stress [53], regulate apoptosis of PDLCs [54] and increase the severity of periodontal inflammation [55]. Inhibits the expression of PPP1R1B and subsequent degradation of the p53 protein in pancreatic cancer cells [56]. Loss of HIF1A can also increase cancer cell proliferation, invasion, and metastasis activity [57]. Transcription factor P53 (tumor protein 53) controls the cell cycle, apoptosis, and cell senescence of periodontal ligament fibroblasts in periodontitis [58]. It plays an important role as a star transcription factor in oral squamous cell carcinoma [59]. Its protein level and phosphorylated protein levels are important factors in suppressing cancer. Low levels of p53 are directly related to the incidence and poor prognosis of oral squamous cell carcinoma [60]. E2F1 (recombinant E2F transcription factor 1) is related to changes in cell metabolism, cell–matrix interaction, and cell cycle [61], and it plays a crucial role in the NF-κB pathway in infection, inflammation and carcinogenesis [62], which can inhibit cell proliferation, migration, invasion and EMT processes. MYCN (N-Myc proto-oncogene protein) is a key marker for cell survival and a key transcription factor for maintaining the homeostasis of the periodontal epithelial barrier and inhibiting periodontal inflammation [63]. Its low expression can promote antiapoptotic resistance and EMT [64]. MYCN is associated with the Wnt/β-catenin pathway in OSCC tumorigenesis and inhibits epithelial-mesenchymal transformation, migration, and colony formation in OSCC. JUN (JUN proto-oncogene protein, AP-1 transcription factor) is related to immune infiltration [65], which causes inflammation and cell death through immunosuppression, leading to cancer.
Functional enrichment analysis of Co-DEmiRNA-Gene, Hub genes and TF networks showed that many cancer-related KEGG/Reactome pathways are enriched, supporting previous findings that PD is a significant risk factor for OSCC (Like PI3 K-related signaling pathway and MAPK pathway). Ras protein signal transduction and the functional enrichment of transcription factor binding in GO analysis are very obvious. These play a crucial role in inflammation, immunosuppression, and antitumor immunity [66,67,68,69].
In this study, the compounds most closely related to Co-DEmiRNA of the two diseases were also analyzed. 5-fluorouracil(5-FU), Ginsenoside, Rh2, and Formaldehyde are the small molecule compounds with the strongest correlation with Co-DEmiRNA. miRNAs reduce the resistance of oral squamous cell carcinoma cells to 5-fluorouracil [70]. At the same time, 5-FU also increases the severity and duration of periodontitis and damages tissue repair by reducing cell and blood vessel renewal, leading to more severe periodontal damage [71]. Ginsenoside Rh2 can control inflammation by regulating the STAT3 signaling pathway and NF-κB signaling pathway to reduce the production of inflammatory factors at mucosal sites [72, 73]. At the same time, it can also inhibit tumor invasion, migration, and angiogenesis by regulating miRNA or AMPK/mTOR and other signaling pathways [74, 75], and induce cancer cell apoptosis and protective autophagy[76]. Formaldehyde is a typical risk factor, which can cause oxidative damage, inflammation, and genotoxicity, and greatly increase the risk of cancer [77, 78]. Future studies will be necessary to investigate these rich compounds in the context of OSCC and PD association.
This study investigated the epigenetic mechanism linked between OSCC and PD, including multiple aspects, such as DEmiRNA, Co-DEmiRNA, Hub gene, TF, and even related compounds. The main limitation is the lack of further experimental data to validate these candidate key linking mechanisms. The datasets used in this study were from a single database, which may limit the accuracy of the results. Future-related research using diverse composite data is critical and necessary. Another point is that other noncoding RNAs, such as lncRNAs, circRNAs, and sncRNAs, may also play an important role in the pathogenic mechanism of OSCC and PD, which were not investigated in this study. Therefore, future studies may further investigate other noncoding RNAs as linkage mechanisms. Future studies should aim to validate the further link between Co-DEmiRNA Hub genes, TF pathway, and compound, these key parts between OSCC and PD, using clinical studies, in vitro and in vivo experiments, etc. In addition, since this association may be bidirectional, it is necessary to comprehensively study the biological mechanisms involved, which will also provide a basis for us to explain the inflammation-cancer transformation further.
Conclusions
Comprehensive analysis of Co-DEmiRNAs in OSCC and PD revealed key genetic molecular mechanisms (Table 5), including miRNAs (including hsa-mir-224, hsa-mir-210, hsa-mir-497, hsa-mir-29c, hsa-mir-486-5p and hsa-mir-31), genes (including ZNF460, FBN1, CDK6, BTG2, CBX6) and TFs (including HIF1A, TP53, E2F1, MYCN, and JUN). It highlighted the most DEmiRNA-related small molecule compounds (including 5-fluorouracil, Ginsenoside Rh2, Formaldehyde and Cisplatin, and Vincristine). These findings provide a theoretical basis to guide future experimental research.
Abbreviations
- OSCC:
-
Oral squamous cell carcinoma
- PD:
-
Periodontitis
- DEmiRNA:
-
Differentially expressed miRNA
- Co-DEmiRNA:
-
Common (co-over expression/co-low expression) DEmiRNA
- TF:
-
Transcription factor
- GO:
-
Gene ontology
- CC:
-
Cellular component
- MF:
-
Molecular function
- BP:
-
Biological process
- EMT:
-
Epithelial-mesenchymal transition
References
Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries (vol 68, pg 394, 2018). Ca-a Cancer J Clinic. 2020;70(4):313–313.
Poveda-Roda R, Bagan JV, Jimenez-Soriano Y, Margaix-Munoz M, Sarrion-Perez G. Changes in smoking habit among patients with a history of oral squamous cell carcinoma (OSCC). Medicina Oral Patologia Oral Y Cirugia Bucal. 2010;15(5):E721–6.
Adeoye J, Tan JY, Ip CM, Choi SW, Thomson P. “Fact or fiction?”: Oral cavity cancer in nonsmoking, nonalcohol drinking patients as a distinct entity-scoping review. Head Neck-J Sci Special Head Neck. 2021;43(11):3662–80.
Rai AK, Freddy AJ, Chatterjee A. Raw betel-nut induced oral and esophageal cancers in Meghalaya. Oral Oncol. 2009;3:206.
Laprise C, Shahul HP, Madathil SA, et al. Periodontal diseases and risk of oral cancer in Southern India: results from the HeNCe Life study. Int J Cancer. 2016;139(7):1512–9.
Shin KH, Park KH, Hong HJ, et al. Prevalence of microsatellite instability, inactivation of mismatch repair genes, p53 mutation, and human papillomavirus infection in Korean oral cancer patients. Int J Oncol. 2002;21(2):297–302.
Ortiz AP, Gonzalez D, Ramos J, et al. Association of marijuana use with oral HPV infection and periodontitis among Hispanic adults: Implications for oral cancer prevention. J Periodontol. 2018;89(5):540–8.
Shin YJ, Choung HW, Lee JH, Rhyu IC, Kim HD. Association of periodontitis with oral cancer: a case-control study. J Dent Res. 2019;98(5):526–33.
Gonda K, Shibata M, Suzuki S, et al. Serum levels of vascular endothelial growth factor in patients with gastrointestinal cancers and correlation with malnutrition, immunosuppression involving MDSC, and systemic inflammation. J Clin Oncol. 2014;32(3):1682.
Shibata M, Ujiie D, Ashizawa M, et al. Correlation of inflammation-related markers with MDSC and IL-17, and use as prognostic indicators in patients with advanced gastric and colorectal cancers. J Clin Oncol. 2019;37(15):e14204.
Gundogdu UD, Cenikh U, Coban FK, Tanriverdi O. Correlation of inflammation parameters with peripheral neuropathic pain in breast cancer patients underwent through paclitaxel chemotherapy. Fresenius Environ Bull. 2021;30(7a):9547–50.
Contaldo M, Boccellino M, Zannini G, et al. Sex hormones and inflammation role in oral cancer progression: a molecular and biological point of view. J Oncol. 2020.
Sfreddo CS, Maier J, De David SC, Susin C, Moreira CHC. Periodontitis and breast cancer: a case-control study. Commun Dent Oral Epidemiol. 2017;45(6):545–51.
Miskiewicz A, Szparecki G, Durlik M, et al. The Q705K and F359L single-nucleotide polymorphisms of NOD-like receptor signaling pathway: association with chronic pancreatitis, pancreatic cancer, and periodontitis. Arch Immunol Ther Exp. 2015;63(6):485–94.
Li X, Chaouhan HS, Li CH, et al. Higher risk of gastric helicobacter pylori infection in patients with periodontitis: a nationwide population-based retrospective cohort study in Taiwan. Int J Environ Res Public Health. 2021;18(21):11678.
Di Spirito F, Toti P, Pilone V, et al. The association between periodontitis and human colorectal cancer: genetic and pathogenic linkage. Life-Basel. 2020;10(9):211.
Komlos G, Csurgay K, Horvath F, Pelyhe L, Nemeth Z. Periodontitis as a risk for oral cancer: a case-control study. BMC Oral Health. 2021;21(1):640.
Javed F, Warnakulasuriya S. Is there a relationship between periodontal disease and oral cancer? A systematic review of currently available evidence. Crit Rev Oncol Hematol. 2016;97:197–205.
Wei L, Li SL, Zhang PC, et al. Characterizing microRNA-mediated modulation of gene expression noise and its effect on synthetic gene circuits. Cell Rep. 2021;36(8):109573.
Chen HW, Lai YC, Rahman MM, et al. Micro RNA differential expression profile in canine mammary gland tumor by next generation sequencing. Gene. 2022;818.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30.
Amr KS, Atia HA, Elbnhawy RA, Ezzat WM. Early diagnostic evaluation of miR-122 and miR-224 as biomarkers for hepatocellular carcinoma. Genes Diseases. 2017;4(4):215–21.
Fan JQ, Xu GX, Chang ZB, Zhu L, Yao J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin Sci. 2020;134(7):807–25.
Dickson MC, Ludbrook VJ, Perry HC, et al. A model of skin inflammation in humans leads to a rapid and reproducible increase in the interferon response signature: a potential translational model for drug development. Inflamm Res. 2015;64(3–4):171–83.
Li CH, Zhu ZY, Hou QS, et al. Revealing potential immunotherapy targets through analysis of a ceRNA network in human colon adenocarcinoma. Transl Cancer Res. 2021;10(12):5319–36.
Wijaya RM, David A. microRNA identification on head and neck squamous cell carcinoma cancer gene based on male gender. Res J Biotechnol. 2021;16(8):50–4.
Denkceken T, Pala E. Investigation of key miRNAs and potential mechanisms in non-small cell lung cancer development from chronic obstructive pulmonary disease. Gen Physiol Biophys. 2020;39(1):69–77.
Tian LL, Qian B, Jiang XH, et al. MicroRNA-497-5p is downregulated in hepatocellular carcinoma and associated with tumorigenesis and poor prognosis in patients. Int J Genom. 2021.
Jiang AM, Liu N, Bai SH, et al. The construction and analysis of tumor-infiltrating immune cells and ceRNA networks in bladder cancer. Front Genet. 2020;11.
Zhang SW, Yue WX, Xie Y, et al. The four-microRNA signature identified by bioinformatics analysis predicts the prognosis of nasopharyngeal carcinoma patients. Oncol Rep. 2019;42(5):1767–80.
Chen T, Zhu JJ, Cai TT, et al. Suppression of non-small cell lung cancer migration and invasion by hsa-miR-486-5p via the TGF-beta/SMAD2 signaling pathway. J Cancer. 2019;10(24):6014–24.
Monterde-Cruz L, Ramirez-Salazar EG, Rico-Martinez G, et al. MicroRNA expression in relation with clinical evolution of osteosarcoma. Pathol Res Pract. 2020;216(8):153038.
Staedel C, Darfeuille F. MicroRNAs and bacterial infection. Cell Microbiol. 2013;15(9):1496–507.
D. D. Niu, Y. E. Lii, P. Chellappan et al., "miRNA863–3p sequentially targets negative immune regulator ARLPKs and positive regulator SERRATE upon bacterial infection," Nature Communications, vol. 7, 2016.
K. L. Menard, B. E. Haskins, and E. Y. Denkers, "Impact of Toxoplasma gondii Infection on Host Non-coding RNA Responses," Frontiers in Cellular and Infection Microbiology, vol. 9, 2019.
Y. S. Chen, J. Li, R. Menon et al., "Dietary spinach reshapes the gut microbiome in an Apc-mutant genetic background: mechanistic insights from integrated multi-omics," Gut Microbes, vol. 13, no. 1, 2021.
Y. Liu, Z. R. Li, Y. X. Qi, X. T. Wen, and L. Zhang, "Metagenomic Analysis Reveals a Changing Microbiome Associated With the Depth of Invasion of Oral Squamous Cell Carcinoma," Frontiers in Microbiology, vol. 13, 2022.
Y. X. Chen, Z. J. Huang, Z. M. Tang et al., "More Than Just a Periodontal Pathogen -the Research Progress on Fusobacterium nucleatum," Frontiers in Cellular and Infection Microbiology, vol. 12, 2022.
Hao TF, Xu JN, Fang SF, et al. Overexpression of ZNF460 predicts worse survival and promotes metastasis through JAK2/STAT3 signaling pathway in patient with colon cancer. J Cancer. 2021;12(11):3198–208.
Zou C, Li X, Lv XZ, et al. Circular RNA mitochondrial translation optimization 1 homologue (CircMTO1) induced by zinc finger protein 460 (ZNF460) promotes oral squamous cell carcinoma progression through the microRNA miR-320a/alpha thalassemia/mental retardation, X-linked (ATRX) axis. Bioengineered. 2021;12(2):9585–97.
Pan MC, Li LJ, Li ZH, et al. Rare variants and polymorphisms of FBN1 gene May increase the risk of non-syndromic aortic dissection. Front Genet. 2022;13:778806.
Kono K, Maeda H, Fujii S, et al. Exposure to transforming growth factor-beta 1 after basic fibroblast growth factor promotes the fibroblastic differentiation of human periodontal ligament stem/progenitor cell lines. Cell Tissue Res. 2013;352(2):249–63.
Yang DY, Zhao DQ, Chen XR. MiR-133b inhibits proliferation and invasion of gastric cancer cells by up-regulating FBN1 expression. Cancer Biomark. 2017;19(4):425–36.
Botticelli A, Fabbri A, Roberto M, et al. The role of the CDK4/6 inhibitor ribociclib in locally advanced and oligometastatic hormone receptor positive, Her2 Negative, advanced breast cancer: case series and review of the literature. Front Oncol. 2022;12:797157.
Sharifi MN, Anandan A, Grogan P, O’Regan RM. Therapy after cyclin-dependent kinase inhibition in metastatic hormone receptor-positive breast cancer: resistance mechanisms and novel treatment strategies. Cancer. 2020;126(15):3400–16.
Petroni G, Formenti SC, Chen-Kiang S, Galluzzi L. Immunomodulation by anticancer cell cycle inhibitors. Nat Rev Immunol. 2020;20(11):669–79.
Wang X, Wang Y. LncRNA DCST1-AS1 inhibits PDLCs’ proliferation in periodontitis and may bind with miR-21 precursor to upregulate PLAP-1. J Periodontal Res. 2020;56(21):256.
Mao BJ, Zhang ZM, Wang G. BTG2: A rising star of tumor suppressors. Int J Oncol. 2015;46(2):459–64.
Yuniati L, Scheijen B, van der Meer LT, van Leeuwen FN. Tumor suppressors BTG1 and BTG2: beyond growth control. J Cell Physiol. 2019;234(5):5379–89.
Yang X, Morita M, Wang H, et al. Crystal structures of human BTG2 and mouse TIS21 involved in suppression of CAF1 deadenylase activity. Nucleic Acids Res. 2008;36(21):6872–81.
Tsui KH, Lin YH, Chung LC, et al. Prostate-derived ets factor represses tumorigenesis and modulates epithelial-to-mesenchymal transition in bladder carcinoma cells. Cancer Lett. 2016;375(1):142–51.
Wang JM, He H, Jiang QC, Wang Y, Jia SZ. CBX6 promotes HCC metastasis via transcription factors Snail/Zeb1-mediated EMT mechanism. Onco Targets Ther. 2020;13:12489–500.
Chen DR, Wu LP, Liu L, et al. Comparison of HIF1A-AS1 and HIF1A-AS2 in regulating HIF-1 alpha and the osteogenic differentiation of PDLCs under hypoxia. Int J Mol Med. 2017;40(5):1529–36.
Kim YS, Shin SI, Kang KL, et al. Nicotine and lipopolysaccharide stimulate the production of MMPs and prostaglandin E-2 by hypoxia-inducible factor-1 alpha up-regulation in human periodontal ligament cells. J Periodontal Res. 2012;47(6):719–28.
Lin YF, Cheng TF, Zhu SY, et al. mRNA and long non-coding RNA expression profiling of human periodontal ligament cells under tension loading. Eur J Orthod. 2021;43(6):698–707.
Tiwari A, Tashiro K, Dixit A, et al. Loss of HIF1A from pancreatic cancer cells increases expression of PPP1R1B and degradation of p53 to promote invasion and metastasis. Gastroenterology. 2020;159(5):1882.
Rohwer N, Bindel F, Grimm C, et al. Annexin A1 sustains tumor metabolism and cellular proliferation upon stable loss of HIF1A. Oncotarget. 2016;7(6):6693–710.
Ambili R, Santhi WS, Janam P, Nandakumar K, Pillai MR. Expression of activated transcription factor nuclear Factor-kappa B in periodontally diseased tissues. J Periodontol. 2005;76(7):1148–53.
Nakazawa S, Sakata K, Liang S, et al. Dominant-negative p53 mutant R248Q increases the motile and invasive activities of oral squamous cell carcinoma cells. Biomed Res-Tokyo. 2019;40(1):37–49.
Sabit H, Tombuloglu H, Cevik E, et al. Knockdown of c-MYC controls the proliferation of oral squamous cell carcinoma cells in vitro via dynamic regulation of key apoptotic marker genes. Int J Mol Cell Med. 2021;10(1):45–55.
Galea GL, Meakin LB, Harris MA, et al. Old age and the associated impairment of bones’ adaptation to loading are associated with transcriptomic changes in cellular metabolism, cell-matrix interactions and the cell cycle. Gene. 2017;599:36–52.
Shi WF, Hou XL, Li X, et al. Differential gene expressions of the MAPK signaling pathway in enterovirus 71-infected rhabdomyosarcoma cells. Braz J Infect Dis. 2013;17(4):410–7.
Dieterle MP, Husari A, Steinberg T, et al. Role of mechanotransduction in periodontal homeostasis and disease. J Dent Res. 2021;100(11):1210–9.
Zhao YC, Wang Y, Chen W, et al. Targeted intervention of eIF4A1 inhibits EMT and metastasis of pancreatic cancer cells via c-MYC/miR-9 signaling. Cancer Cell Int. 2021;21(1):670.
Ye F, Li JT, Xu PT, et al. Osteogenic differentiation of mesenchymal stem cells promotes c-Jun-dependent secretion of interleukin 8 and mediates the migration and differentiation of CD4(+) T cells. Stem Cell Res Therapy. 2022;13(1):58.
Sun Y, Zhao Y, Wang XP, et al. Wogonoside prevents colitis-associated colorectal carcinogenesis and colon cancer progression in inflammation-related microenvironment via inhibiting NF-kappa B activation through PI3K/Akt pathway. Oncotarget. 2016;7(23):34300–15.
Seshadri VD. Brucine promotes apoptosis in cervical cancer cells (ME-180) via suppression of inflammation and cell proliferation by regulating PI3K/AKT/mTOR signaling pathway. Environ Toxicol. 2021;36(9):1841–7.
Li S, Zhang Y, Gao AS, Zhang Y, Zhang J. Engelharquinone suppresses lipopolysaccharide-induced inflammation and proliferation of human liver cancer SMCC7721 cells via inhibition of NF-kappa B and MAPK signaling pathway. Trop J Pharm Res. 2020;19(4):699–706.
Vahed SZ, Barzegari A, Saadat YR, Goreyshi A, Omidi Y. Leuconostoc mesenteroides-derived anticancer pharmaceuticals hinder inflammation and cell survival in colon cancer cells by modulating NF-kappa B/AKT/PTEN/MAPK pathways. Biomed Pharmacother. 2017;94:1094–100.
Liu BW, Hu JC, Zhao H, Zhao L, Pan SY. MicroRNA-155-5p contributes to 5-fluorouracil resistance through down-regulating TP53INP1 in oral squamous cell carcinoma. Front Oncol. 2022;11:706095.
Novaes VCN, Ervolino E, Fernandes GL, et al. Influence of the treatment with the antineoplastic agents 5-fluorouracil and cisplatin on the severity of experimental periodontitis in rats. Support Care Cancer. 2022;30(3):1967–80.
Bi WY, Fu BD, Shen HQ, et al. Sulfated derivative of 20(S)-ginsenoside Rh2 inhibits inflammatory cytokines through MAPKs and NF-kappa B pathways in LPS-induced RAW264.7 macrophages. Inflammation. 2012;35(5):1659–68.
Wang YS, Chen C, Zhang SY, Li Y, Jin YH. (20S) Ginsenoside Rh2 inhibits STAT3/VEGF signaling by targeting annexin A2. Int J Mol Sci. 2021;22(17):9289.
Jiang S, Yan JQ, Chen XS, et al. Ginsenoside Rh2 inhibits thyroid cancer cell migration and proliferation via activation of miR-524-5p. Arch Med Sci. 2021;18(1):164–70.
Li CC, Gao H, Feng XM, et al. Ginsenoside Rh2 impedes proliferation and migration and induces apoptosis by regulating NF-kappa B, MAPK, and PI3K/Akt/mTOR signaling pathways in osteosarcoma cells. J Biochem Mol Toxicol. 2020;34(12):e22597.
Bian S, Zhao Y, Li FY, et al. Knockdown of p62/sequestosome enhances ginsenoside Rh2-induced apoptosis in cervical cancer HeLa cells with no effect on autophagy. Biosci Biotechnol Biochem. 2021;85(5):1097–103.
Vishnu US, Esteve PO, Chin HG, Pradhan S. One-pot universal NicE-seq: all enzymatic downstream processing of 4% formaldehyde crosslinked cells for chromatin accessibility genomics. Epigenet Chrom. 2021;14(1):58.
Protano C, Buomprisco G, Cammalleri V, et al. The carcinogenic effects of formaldehyde occupational exposure: a systematic review. Cancers. 2022;14(1):165.
Acknowledgements
We would like to express our deep gratitude to our colleagues for their contributions.
Funding
The work was supported by the National Natural Science Foundation of China (81771127), the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01C15), the Medical Engineering Cross Foundation of Shanghai Jiao Tong University (YG2015MS06), and the Seed Foundation of the Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (JYZZ196).
Author information
Authors and Affiliations
Contributions
Conceptualization: ZL; Methodology: ZL, RF and XW; Data Acquisition: RF; Formal Analysis: RF; Manuscript preparation: ZL; Review and Edit: ZL, XW; Supervision: LZ; Revision: LZ. All authors have read and agreed to the published version of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The datasets analyzed during the current study are available in the GEO repository. (https://www.ncbi.nlm.nih.gov/geo/).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1a.
Differentially Expressed miRNA in Oral Squamous Cell Carcinoma (GSE45238). Table S1b. Differentially expressed miRNA in Periodontitis (GSE54710). Table S2a. Co-DEmiRNA-Gene Minimum Network. Table S2b. KEGG Pathway Enrichment Analysis of Co-DEmiRNA-Gene Network. Table S2c. Reactome Pathway Enrichment Analysis of Co-DEmiRNA-Gene Network. Table S2d. Gene Ontology-Biological Process Enrichment Analysis of Co-DEmiRNA-Gene Network. Table S2e. Gene Ontology-Molecular Functions Enrichment Analysis of Co-DEmiRNA-Gene Network. Table S2f. Gene Ontology-Cellular Component Enrichment Analysis of Co-DEmiRNA-Gene Network. Table S3. The Transcription Factors of 11 Co-DEmiRNA. Table S4a. Co-DEmiRNA-TF Minimum Network. Table S4b. KEGG Pathway Enrichment Analysis of Co-DEmiRNA-TF Network. Table S4c. Reactome Pathway Enrichment Analysis of Co-DEmiRNA-TF Network. Table S4d. Gene Ontology-Biological Process Enrichment Analysis of Co-DEmiRNA-TF Network. Table S4e. Gene Ontology-Molecular Functions Enrichment Analysis of Co-DEmiRNA-TF Network. Table S4f. Gene Ontology-Cellular Component Enrichment Analysis of Co-DEmiRNA-TF Network. Table S5. Co-DEmiRNA-Compound Minimum Network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Fu, R., Wen, X. et al. Network analysis reveals miRNA crosstalk between periodontitis and oral squamous cell carcinoma. BMC Oral Health 23, 19 (2023). https://doi.org/10.1186/s12903-022-02704-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-022-02704-2