
Abstract
Background
The voltage-gated potassium channel Kv7.1 encoded by KCNQ1 is located in both cardiac myocytes and insulin producing beta cells. Loss-of-function mutations in KCNQ1 causes long QT syndrome along with glucose-stimulated hyperinsulinemia, increased C-peptide and postprandial hypoglycemia. The KCNE1 protein modulates Kv7.1 in cardiac myocytes, but is not expressed in beta cells. Gain-of-function mutations in KCNQ1 and KCNE1 shorten the action potential duration in cardiac myocytes, but their effect on beta cells and insulin secretion is unknown.
Case presentation
Two patients with atrial fibrillation due to gain-of-function mutations in KCNQ1 (R670K) and KCNE1 (G60D) were BMI-, age-, and sex-matched to six control participants and underwent a 6-h oral glucose tolerance test (OGTT). During the OGTT, the KCNQ1 gain-of-function mutation carrier had 86% lower C-peptide response after glucose stimulation compared with matched control participants (iAUC360min = 34 pmol/l*min VS iAUC360min = 246 ± 71 pmol/l*min). The KCNE1 gain-of-function mutation carrier had normal C-peptide levels.
Conclusions
This case story presents a patient with a gain-of-function mutation KCNQ1 R670K with low glucose-stimulated C-peptide secretion, additionally suggesting involvement of the voltage-gated potassium channel KCNQ1 in glucose-stimulated insulin regulation.
Similar content being viewed by others

Background
Impaired function of the voltage-gated potassium channels Kv7.1 (encoded by KCNQ1) and Kv11.1 (encoded by KCNH2), caused by inheritable mutations or drugs leads to long QT syndrome (LQTS) characterised by malignant cardiac arrhythmias [1]. Moreover, inhibition of Kv7.1 and Kv11.1 increases the glucose-stimulated insulin and C-peptide secretion from the pancreatic beta cells and increases glucagon-like peptide (GLP)-1 in mice [2, 3] and we have previously shown that patients with LQT1 (due to loss of function mutations in KCNQ1) and LQT2 (due to loss of function mutations in KCNH2) have glucose-stimulated hyperinsulinemia and postprandial hypoglycaemia [2, 4].
KCNQ1 is expressed in human beta cells [5] and blockage of the channel increases glucose- stimulated insulin secretion [3], and its overexpression impairs glucose-stimulated insulin secretion [6]. KCNE1 encodes a human potassium channel accessory (β) subunit, and modulates Kv7.1 in cardiomyocytes, but does not seem to be expressed in beta cells [7]. Gain-of-function mutations in either KCNQ1 or KCNE1 genes shorten the action potential duration and effective refractory period in cardiomyocytes, increasing the risk of atrial fibrillation (AF) [8, 9].
In this case study, we investigated glucose-stimulated hormone secretion in two patients with AF due to confirmed gain-of-function mutations KCNQ1 R670K and KCNE1 G60D, respectively. Expression in Xenopus laevis oocytes of KCNQ1 R670K or Kv7.1 co-expressed with KCNE1 G60D resulted in larger current amplitudes compared with wildtype, confirming a gain-of-function phenotype [8, 9] of the mutations.
We hypothesized that patients with a KCNQ1 gain of function mutation would have decreased glucose-induced insulin and C-peptide secretion, whereas patients with gain of function mutations in KCNE1 would be expected to have normal insulin and C-peptide secretion upon glucose stimulation.
Case presentation
We present two patients with AF who are confirmed heterozygous gain-of-function mutations carriers, recruited from the outpatient clinic at Department of Cardiology, Rigshospitalet, Denmark. One patient had persistent AF and carried the KCNQ1 R670K mutation, while the other patient had paroxysmal AF and carried the KCNE1 G60D mutation. Neither patients had echocardiography abnormalities. For comparison with normal glucose metabolism and ECG profiles, six control participants were BMI, age and sex-matched with the AF patients recruited from the Danish populations studies Inter99, Health 2006, Health 2010 and DanFund studies.
The methods used for the investigations and sample analyzing were previously detalied described in [2]. Below follows a condensed version. The patients and control participants each underwent a 6-h oral glucose tolerance test (OGGT) after overnight fasting. The patients did not take medication the morning before the examination. In a resting state, baseline ECG and blood samples were taken 15, 10 and 0 min before ingestion of a standard 75 g glucose solution. During the following 6 h, ECG and blood samples were taken every 15 min for the first hour and then every 30 min for the remaining 5 h.
Height and weight were measured and BMI calculated as height (m) / weight (kg)^2. Fat percentage was measured using bioimpedance (Biodynamics BIA 310e, Biodynamics, Seattle, WA).
Plasma glucose was measured using an automated Vitros 5.1 FS/5600 analyzer (Ortho Clinical Diagnostics, lower quantitation limit: 19.8 mg/Dl, intra- and interassay coefficients of variation: 0.025). Serum C-peptide was measured using an automated Cobas e411 analyzer (Roche) (analytic detection limit: 1.4–3 pmol/L, intra- and interassay coefficients of variation < 0.04 and < 0.025 respectively. Plasma glucagon and GLP-1 were measured using validated radioimmunoassays with a detection limit < 1 pmol/L [10].
12-lead ECGs were recorded in a resting supine position using a MAC1600 ECG machine (GE Healthcare, Milwaukee, WI). Bazett’s formula (QTcB = QT/(RR)1/2) and Fridericia’s formula (QTcF = QT/(RR)1/3) were used to correct the QT interval by heart rate (RR).
For continuous glucose monitoring (CGM), the participants agreed to wear an iPro2 CGM (Medtronic, Watford, U.K.) between 3 and 7 days. During this period each meal was noted with time and meal composition.
HOMA-IR index was calculated as (fasting glucose (mmol/l) x fasting insulin (μIU/ml))/22.5. HOMA-Beta was calculated (20 x fasting insulin (μIU/ml))/(fasting glucose (mmol/ml) - 3.5).
Findings: There were no differences in HbA1c, fasting hemoglobin, fasting total cholesterol or fasting creatinine between the patients and the corresponding control participants. None of them had HbA1c levels ≥48 mmol/mol (Table 1).
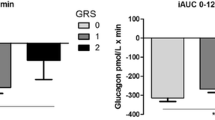
At fasting state, the KCNQ1 R670K carrier presents with slightly higher fasting insulin levels (but still within the levels observed in the control participants (insulin 88 vs range 14–137 pmol/L and C-peptide 774 vs 338–1226 pmol/L) and therefore increased HOMA-IR (3.1 vs 1.5 ± 1.2) and HOMA-Beta (123% vs 70 ± 55% compared to control participants). In contrast, during glucose stimulation the KCNQ1 R670K carrier had a markedly blunted C-peptide response and lower glucose levels compared to control participants and the KCNE1 G60D carrier (Fig. 1 and S1). The glucose-stimulated GLP-1 response was also blunted in the KCNQ1 GOF patient compared to control participants, whereas glucagon response did not differ among the examined participants (Fig. 1). During CGM for 3–7 days, the KCNQ1 mutation carrier had lower increase in blood glucose levels within 1 h after carbohydrate rich meals (mean increase of 0.8 ± 0.6 mmol/l), compared to both matched controls and the KCNE1 mutation carrier (mean increase of 1.5 ± 0.4 mmol/L and 1.7 ± 0.5 mmol/L, respectively) (Fig. S2).

Plasma glucose and hormone responses to oral glucose tolerance test (OGTT). Six hour OGTT and results from Glucose (a), C-peptide (b), Glucagon (c), and GLP-1 (d) of the KCNQ1 R670K carrier (KCNQ1) and KCNE1 G60D carrier (KCNE1) and their BMI, sex and age matched control participants (n = 6), mean ± SEM
The two patients had similar cardiac profiles as previously reported [8, 9] (Fig. 2).

Results from the ECG measurement during the oral glucose tolerance test (OGTT). Six hour OGTT and results from Heart rate (a), QTcB (b) and QTcF (c) of the KCNQ1 R670K carrier (KCNQ1) and KCNE1 G60D carrier (KCNE1) and their BMI, sex and age matched control participants (n = 4), means ± SEM
Discussion and conclusions
We previously identified that patients with loss-of-function mutations in KCNQ1 have increased glucose-stimulated C-peptide and insulin secretion, but normal fasting levels [4]. Kv7.1 is expressed in human beta cells and participates in depolarization-evoked insulin exocytosis [5]. In this case study of a patient with gain of function mutation in KCNQ1, we observed a lower glucose-stimulated C-peptide and GLP-1 response compared to matched control participants. This may be due to shorter repolarization duration in beta and L-cells cells, similar to what has been observed for this variant with shortened action potential duration in Xenopus laevis oocytes [8, 9]. Furthermore, this observation is in agreement with studies of overexpression of KCNQ1 showing an increased glucose stimulated K+ current and impaired and limited glucose stimulated insulin secretion from beta-cells [6]. The KCNQ1 gain of function patient had a low increase in glucose level after glucose ingestion both during OGTT and during 7 days continuous glucose monitoring, even though the C-peptide in response to glucose stimulation were low. The patient reported that he bicycled more than 20 km every weekday, which may explain the high insulin sensitivity in the glucose-stimulated state of the patient making him able to compensate for the low C-peptide levels by increased glucose uptake in the muscles. Intron variants in KCNQ1 associated with increased risk of type 2 diabetes in genome wide association studies seem to increase the function of KCNQ1, whereas siRNA silencing decrease KCNQ1 function and increase exocytosis of insulin [5].
Thus, with time and without exercise the KCNQ1 gain of function patient may be in risk of type 2 diabetes.
We examined another AF patient with gain of function mutation in KCNE1 that is not expressed in pancreas [7], this patient had a similar C-peptide response compared to the matched control participants. Hence KCNE1 does not seem to function as a beta subunit of KCNQ1 in human beta-cells.
Although limited by the very modest sample size, this study provides additional suggestions of the involvement of the voltage-gated potassium channel KCNQ1 in insulin regulation.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable.
Abbreviations
- AF:
-
Atrial fibrillation
- LQTS:
-
Long QT syndrome
- OGTT:
-
Oral glucose tolerance test
References
Attali B. Ion channels. A new wave for heart rhythms. Nature. 1996;384(6604):24–5.
Hyltén-Cavallius L, Iepsen EW, Wewer Albrechtsen NJ, Svendstrup M, Lubberding AF, Hartmann B, Jespersen T, Linneberg A, Christiansen M, Vestergaard H. Patients with long-QT syndrome caused by impaired hERG-encoded Kv11. 1 potassium channel have exaggerated endocrine pancreatic and incretin function associated with reactive hypoglycemia. Circulation. 2017;135(18):1705–19.
Liu L, Wang F, Lu H, Ren X, Zou J. Chromanol 293B, an inhibitor of KCNQ1 channels, enhances glucose-stimulated insulin secretion and increases glucagon-like peptide-1 level in mice. Islets. 2014;6(4):e962386.
Torekov SS, Iepsen E, Christiansen M, Linneberg A, Pedersen O, Holst JJ, Kanters JK, Hansen T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes. 2014;63(4):1315–25.
Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, Zhang E, Almgren P, Ladenvall C, Axelsson AS, et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61(7):1726–33.
Yamagata K, Senokuchi T, Lu M, Takemoto M, Fazlul Karim M, Go C, Sato Y, Hatta M, Yoshizawa T, Araki E, et al. Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 beta-cell line. Biochem Biophys Res Commun. 2011;407(3):620–5.
Lundquist AL, Turner CL, Ballester LY, George AL. Expression and transcriptional control of human KCNE genes. Genomics. 2006;87(1):119–28.
Steffensen AB, Refsgaard L, Andersen MN, Vallet C, Mujezinovic A, Haunso S, Svendsen JH, Olesen SP, Olesen MS, Schmitt N. IKs gain- and loss-of-function in early-onset lone atrial fibrillation. J Cardiovasc Electrophysiol. 2015;26(7):715–23.
Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Medical Genetics. 2012;13:24.
Orskov C, Jeppesen J, Madsbad S, Holst JJ. Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest. 1991;87(2):415–23.
Acknowledgements
We thank the study participants and the technicians Annemette Forman and Lene Albæk.
Funding
The study was supported by the Novo Nordisk Foundation Center for Basic Metabolic Synergy Grant (SST) and the Danish Heart Association (SST). LH-C was supported by a scholarship from the Lundbeck Foundation, and JZ was supported by the scholarship from China Scholarship Council. None of the funders were involved in the planning and conduction of the study, analysis and interpretation of the data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SST, JK, and TH designed the study. MSO provided the patients. AL provided the cohort of matched control participants. LH-C collected data. JZ analyzed and evaluated data. CRJ contributed to evaluating data. JZ wrote the manuscript with help from SST. TH, JJH, AL, MSO, SST, JK and CRJ contributed to discussion, reviewed/edited the manuscript and approved the final version. The corresponding authors JK and SST confirms full access to data and final responsibility for the decision to submit for publication. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Before participation, informed written consent was obtained from all participants. The project was approved by The Committees on Health Research Ethics in the Capital Region of Denmark (reference number: H-4-2010-036) and was performed in accordance to the Helsinki Declaration II. Participation in the investigation was voluntary and the individuals could retract their consent to participate at any time.
Clinical Trial Registration number: NCT02775513. The manuscript adheres to the CARE guidelines.
Consent for publication
Before participation, informed written consent was obtained from all participants, which include consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Plasma glucose and C-peptide responses to oral glucose ingestion in the KCNQ1 R670K carrier (KCNQ1) and KCNE1 G60D carrier (KCNE1) and their BMI, sex and age matched control participants. Control to KCNQ1 (R670K) (n = 2, men, BMI = 26.8 ± 0.7, age = 49.4 ± 2.3, fat% = 23.9 ± 3.5). Control to KCNE1 (G60D) (n = 2, men, BMI = 19.6 ± 1.3, age = 49.4 ± 0.6, fat% = 16.8 ± 6.6). Figure S2. Results from 3 to 7 day continuous glucose monitors (CGM). Increase of blood glucose levels within 1 h after carbohydrate rich meals(a) and the mean glucose levels for during the whole period(b) from the KCNQ1 (red) R670K carrier (KCNQ1) and KCNE1 G60D carrier (KCNE1) (blue) and their matched control participants, means ± SEM.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, J., Juhl, C.R., Hylten-Cavallius, L. et al. Gain-of-function mutation in the voltage-gated potassium channel gene KCNQ1 and glucose-stimulated hypoinsulinemia - case report. BMC Endocr Disord 20, 38 (2020). https://doi.org/10.1186/s12902-020-0513-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-020-0513-x