
Abstract
Background
The thermophilic filamentous fungus Myceliophthora thermophila has many suitable characteristics for industrial biotechnology and could be a promising new chassis system for synthetic biology, particularly the ATCC 42464 strain, whose genome was sequenced in 2011. However, metabolic engineering of this strain using genetic approaches has not been reported owing to a lack of genetic tools for this organism.
Results
In the present study, we developed a high efficiency Agrobacterium tumefaciens mediated transformation system for M. thermophila, including an approach for targeted gene deletion using green fluorescence protein (GFP) as a marker for selection. Up to 145 transformants per 105 conidia were obtained in one transformation plate. Moreover, a ku70 deletion mutant was constructed in the ATCC 42464 background using the tools developed in present study and subsequently characterized. The ku70 deletion construct was designed using resistance to phosphinothricin as the selection marker. Additionally, a GFP-encoding cassette was incorporated that allowed for the selection of site-specific (no fluorescence) or ectopic (fluorescence) integration of the ku70 construct. Transformants with ectopically integrated ku70 deletion constructs were therefore identified using the fluorescent signal of GFP. PCR and Southern blotting analyses of non-fluorescent putative ku70 deletion transformants revealed all 11 tested transformants to be correct deletions. The deletion frequency in a pool of 116 transformants analyzed was 58 %. Moreover, the homologous rate improved about 3 folds under ku70 mutant using the pyrG as a test gene to disrupt in M. thermophila.
Conclusions
We successfully developed an efficient transformation and target gene disruption approach for M. thermophila ATCC 42464 mediated by A. tumefaciens. The tools and the ku70 deletion strain developed here should advance the development of M. thermophila as an industrial host through metabolic engineering and accelerate the elucidation of the mechanism of rapid cellulose degradation in this thermophilic fungus.
Similar content being viewed by others

Background
The thermophilic filamentous fungus Myceliophthora thermophila exhibits a large capacity for biomass degradation and represents a potential reservoir of novel enzymes for industrial applications, including abundant thermostable enzymes for biomass degradation [1–5]. M. thermophila also has the potential to be a cell factory to produce chemicals and biofuels from renewable lignocellulose [5]. Therefore, metabolic engineering of M. thermophila is attractive. To use M. thermophila in industrial biotechnology, the development of versatile genetic tools is urgently needed.
PEG-mediated protoplast transformation was developed to knock out target genes and for hetero-expression of interesting genes in several filamentous fungi, such as Aspergillus and Trichoderma [6–8]. It was previously reported this approach can be used to transform the industrial strain C1, previously known as Chrysosporium lucknowense, and recently renamed as an isolate of Myceliophthora thermophila [9]. However, preparation of fungal protoplasts was laborious, and heterokaryotic transformants might have genetic stability concerns [10, 11]. The C1 strain is a patented strain, therefore the ATCC 42464 strain is predominantly used for public research as the general wild-type strain of M. thermophila. The whole genome sequence of ATCC 42464 has been completed and is open to the public [12]. Various studies have been performed with M. thermophila ATCC 42464, such as biochemical characterization of enzymes [13–15] and investigating cellulose degradation on a cellular and genomic level [4, 12, 16, 17]. However, molecular engineering using a genetic approach has not been reported in M. thermophila ATCC 42464 because of a lack of mature genetic tools. Agrobacterium tumefaciens-mediated transformation has become a common technique for filamentous fungi [18–22]. This technique allows transfer of a segment from its Ti plasmid into plant or fungi cells, and the T-DNA integrates into the nuclear chromosome either randomly or specifically. The direct transformation of conidia makes A. tumefaciens T-DNA transfer a powerful and easy to use tool for genetic transformation of filamentous fungi. A variety of fungi have been successfully transformed by this approach [18–22]. However, the application of this approach in thermophilic filamentous fungi, including M. thermophila, has not been tested so far.
In present study, we developed a high efficiency transformation and targeted gene disruption system mediated by A. tumefaciens for M. thermophila ATCC 42464. Using this technique, up to 145 transformants were obtained per transformation plate, and a ku70 deletion strain was constructed in the ATCC 42464 background and characterized. All of the tools and strains developed here should facilitate the development of M. thermophila metabolic engineering and accelerate the elucidation of the fascinating cellulose degradation mechanism in this thermophilic fungus.
Results and discussion
Marker selection and genetic transformation in the ATCC 42464 strain
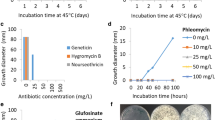
To perform M. thermophila transformation mediated by A. tumefaciens, the sensitivity of this fungus to hygromycin B (Amresco, Solon, OH, USA) and phosphinothricin (Sigma-Aldrich, St. Louis, MO, USA) was tested. Five concentrations of two antibiotics were used. The mature conidia were harvested in 0.05 % Tween 80, and 103 conidia were spread on MM (Vogel’s minimal medium supplemented with 2 % sucrose) agar plates supplemented with various concentrations of antibiotic (Hygromycin B: 25, 50, 75, 100, 150 μg/mL; phosphinothricin: 25, 50, 100, 150, 200 μg/mL). After incubation at 45 °C for 5 days, Hygromycin B showed little inhibition for M. thermophila at more than 100 ug/mL, whereas phosphinothricin concentration completely inhibited the growth of this fungus at 100 ug/mL. Thus, the bar gene conferring fungal resistance to phosphinothricin was employed as the selection marker for M. thermophila transformation.
The Ti vector pPK2BarGFPD containing the trpC promoter from Aspergillus nidulans and the tef promoter from Aureobasidium pullulans (PtrpC and Ptef, respectively) developed in a previous study [23] was chosen as the test plasmid for genetic transformation in M. thermophila ATCC 42464. The vector contains a GFP reporter gene as well as a bar selection marker gene, which confers resistance to phosphinothricin. PCR analysis of the transformants showed the T-DNA was integrated in the chromosomes. High levels of green fluorescent protein (GFP) signal were clearly detected in the conidia and mycelia of transformants (Fig. 1), whereas no autofluorescent signal was observed in the parental strain. This suggested pPK2BarGFPD and its elements, the promoters and the GFP gene, were functioning and could be used for gene over-expression and protein localization analysis in M. thermophila ATCC 42464.

Confocal fluorescence imaging of mycelia (Top) and conidia (Bottom) of M. thermophila pPK2BarGFPD transformants
Optimization of M. thermophila transformation mediated by A. tumefaciens
In an attempt to develop a simple, high efficiency transformation system mediated by A. tumefaciens in M. thermophila ATCC 42464, we performed transformation optimization as described previously [21]. Acetosyringone (AS) serves as an inducer of virulence (vir) genes, whose expression is a prerequisite for A. tumefaciens T-DNA transfer. AS concentration influences the frequency of A. tumefaciens-mediated transformation for Saccharomyces cerevisiae and several filamentous fungi [11, 20, 24, 25]. Therefore, the impact of AS on this approach for M. thermophila was first investigated. As shown in Fig. 2a, using the binary plasmid pPK2BarGFPD, the maximum transformation efficiency (145 ± 10 transformants per plate) was obtained with AS at a concentration of 200 μM, and the number of transformants was reduced when the concentration was increased or decreased. The observation that the higher concentration of AS resulted in a reduction in transformation efficiency is similar to that for Fusarium avenaceum. However, it is in contrast to that for Beauveria bassiana, where an increase in the concentration from 100 μM to 800 μM resulted in increasing numbers of transformants [21, 26]. Based on these observation, the different fungus seems has different tolerance to Acetosyringone (AS), the low frequency of transformation induced by the increased AS might be attributed to the toxic effect of AS on M. thermophila, and the deficient level of vir gene expression at low AS concentrations might be responsible for poor efficiency of A. tumefaciens T-DNA transfer.

Optimization of A. tumefaciens-mediated M. thermophila transformation. a The impact of acetosyringone (AS) on A. tumefaciens-mediated transformation efficiency. Co-cultivation was conducted in induction medium containing AS on indicated concentrations for 2 days. b The effect of co-culture time on transformation efficiency. Co-cultivation was conducted in induction medium containing 200 μM AS for various times indicated. Error bars indicate the standard deviation of three independent experiments
The transformation efficiency is also dependent on co-culture time of M. thermophila and A. tumefaciens. The largest number of transformants was observed with a co-culture time of 2 days, and alterative co-culture times impaired the transformation efficiency (Fig. 2b). Similar results were obtained in Metarhizium anisopliae, Aspergillus terreus and Beauveria bassiana transformation mediated by A. tumefaciens [2, 18, 27], whereas the co-culture time had no influence on a Ganoderma lucidum transformation system [28]. In conclusion, with the optimized approach, a high transformation efficiency of 145 transformants per 105 conidia was obtained. This was higher compared with the amount of transformants obtained in fungi using other approaches, typically less than 100 transformants per 105 conidia [18, 29–31].
Mitotic stability analysis of transformants
The mitotic stability of the T-DNA in M. thermophila transformants was examined next. Twenty randomly selected transformants containing pPK2BarGFPD were cultured on media without phosphinothricin for five generations. After five generations, 19 transformants (95 %) of 20 tested strains retained the resistance to phosphinothricin. Satisfyingly, PCR analysis showed the presence of bar in the 19 transformants, and GFP signal was observed under the fluorescence microscope.
ku70 disruption of M. thermophila ATCC 42464
The high transformation frequency, together with the precision and simplicity of T-DNA, make A. tumefaciens-mediated transformation a suitable genome mutagenesis approach in filamentous fungi, such as Trichoderma, Aspergillus, Beauveria and Metarhizium [22, 27, 32, 33]. Ku70 and Ku80 make up the Ku heterodimer, which binds to the ends of double-stranded DNA breaks and is required for the non-homologous end joining (NHEJ) pathway of DNA repair [34]. In some fungi, including the M. thermophila C1 strain, Trichoderma reesei and Neurospora crassa, mutation of ku70 results in a dramatically increased homologous integration frequency with short homologous flanks [9, 35, 36]. Therefore, disruption of ku70 of the ATCC 42464 strain was performed to test the efficiency of this approach for knocking out of a specific gene in wild-type M. thermophila.
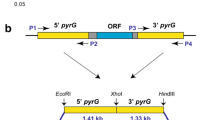
The binary vector pPK2BarGFPD was employed for DNA manipulation and the ku70 knockout cassette (Fig. 3) was constructed as described in the Methods section. The gene encoding green fluorescence protein on the binary vector served as the marker to differentiate between ectopic insertion and correct gene replacement. Fluorescence should not be detected in the transformants where the target gene (ku70) was properly replaced by the bar cassette (Fig. 4a). Conversely, if the fluorescent signal is detected in a transformant, this suggests the bar-gfp segment of the T-DNA was ectopically integrated elsewhere in the genome (random insertion) rather than at the correct gene locus. Using this strategy, we performed the ku70 disruption experiment. One hundred sixteen primary transformants were obtained. Forty-six (39.7 %) transformants were filtered out by an obvious fluorescent signal, indicating they were ectopic insertion mutants. The remaining 70 transformants (60.3 %) with no detectable fluorescent signal under the microscope were likely free of ectopic insertions and passed the first round screening. Subsequent PCR analysis showed the ku70 gene was properly replaced by the bar cassette in 68 of these 70 transformants. To further confirm this observation, we randomly selected eleven PCR-confirmed transformants to perform Southern blotting analysis, and the result showed all 11 tested mutants were correct (Fig. 4b and c). Taken together, these results indicate as many as 58 % (68 of 116) of the transformants could be correct ku70 disruption mutants, indicating a very high rate of homologous recombination. Previous studies reported that the efficiencies of genetic targeted disruption via transformation mediated by A. tumefaciens varied greatly in fungal species, such as 0.04 % for Blastomyces dermatitidis, 29 % for Aspergillus awamori, 74 % for Fusarium avenaceum and 85 % for Fusarium graminearum [37, 38]. Compared with transformation by direct electroporation transformation of protoplasts or conidia, the transformation mediated by A. tumefaciens seems more likely to achieve a higher successful gene disruption rate, at least for some of the species. For example, in Neurospora crassa the homologous recombination rate was usually low (<10 %) without NHEJ disruption using electroporation [35]. Although future studies with more precise statistical analyses are needed to support the improved homologous recombination rate, if validated, it will be a boost for genetic manipulation in this strain, and will speed up strain engineering for industrial applications.

Vector map of the pPK2-ku70 vector constructed based on binary vector pPK2BarGFPD. Kan, kanamycin resistance gene; Ptef, promoter of translation elongation factor gene from Aureobasidium pullulans; egfp, enhanced green fluorescence protein; PtrpC, promoter of tryptophan synthetase gene from Aspergillus nidulans; bar, phosphinothricin resistance gene; 3′-flank and 5′-flank, 3′ and 5′ flanking fragments of ku70, respectively; RB and LB, right and left border of T-DNA, respectively

ku70 disruption of wild-type M. thermophila ATCC 42464. a Pattern of the ku70 knockout cassette integrating into the chromosomes of M. thermophila ATCC 42464 via ectopic insertion or homologous recombination. b PCR analysis of ku70 deletion transformants with one primer (ku70KO-F) located in the bar gene cassette and the other (ku70KO-R) located in the downstream of 3′ flank in the genomic DNA (expected product of 1872 bp) and c Southern blotting analysis of ku70 mutants with XhoI digested genomic DNA and the probe amplified from the 3′ flank. Lines 1–11 in (b) and (c), genomic DNA from ku70 mutants (expected product of 5056 bp). WT, genomic DNA from wild-type as negative control (expected product of 1703 bp). Primers are represented by solid black arrows. Abbreviations: gDNA, genomic DNA; RB and LB, right and left border of T-DNA, respectively
There was no noticeable difference between ku70 disruption mutants and the wild-type strain during our analysis (Fig. 5). After five generations of sub-culturing on MM agar medium, 10 randomly selected ku70 mutants were shown to be mitotically stable. Taken together, these results indicate the approach we developed is an efficient system for gene disruption in M. thermophila ATCC 42464.

Phenotypic characterization of ku70 mutant. a Protein concentration in supernatants from wild-type (WT) and ku70 mutant cultures grown on 1 % Avicel for 3 d at 45 °C. b SDS-PAGE gel of wild-type and Δku70 strains grown at 45 °C for 3 d on 1 % Avicel. c Conidiation of wild-type and Δku70 strains growth at 45 °C for 9 d on agar plate without selective agent. d The hyphae of wild-type and Δku70 strains growth at 45 °C for 24 h on liquid MM without selective agent
In order to check whether the gene homologous recombination rate was improved in ku70 mutant compared to that in wild type strain, we chose the potential marker gene pyrG (MYCTH_2311494) as a test to disrupt in both wild-type and ku70 deletion strain. As expected, 97 % (29/30) of transformants were correct pyrG deletion mutants under ku70 background, whereas only 30 % (12/40) of transformants were correct in wild type background (Fig. 6). This result clearly shown the deletion of KU70 gene can improve the gene homologous recombination in this strain, similar as reported before [9].

pyrG gene deletion in wild type and ∆ku70 mutant of M. thermophila ATCC 42464. The experiment design (a), PCR analysis of transformants under wild-type background (b) and ∆ku70 mutant (c) with one primer (pyrGout-F) located in the neo gene cassette and the other (pyrGout-R) located in the downstream of 3′ flank in the genomic DNA. The genomic DNA from the wild-type (WT) and ∆ku70 strains was used as the negative control
Taken together, these results indicate the approach we developed is an efficient system for gene disruption in M. thermophila ATCC 42464 and will speed up its strain engineering for industrial applications.
Conclusions
We successfully developed a simple and highly efficient transformation approach mediated by A. tumefaciens for the thermophilic filamentous fungus M. thermophila. This study established a targeted gene deletion approach using green fluorescence protein as a selection helper to filter the ectopic insertion. The tools and ku70 mutant developed by the present study should facilitate M. thermophila strain development for industrial applications in the future.
Methods
Strains and culture conditions
Myceliophthora thermophila ATCC 42464 obtained from the American Type Culture Collection (ATCC) was grown on Vogel’s minimal medium supplemented with 2 % sucrose (MM medium) at 45 °C for 15 d to obtain conidia. Sucrose was purchased from Amresco (Solon, OH, USA).
Escherichia coli DH5α was used for plasmid amplification and vector construction and cultivated in Luria–Bertani medium with antibiotics according to conditions for plasmid selection.
Agrobacterium tumefaciens AGL-1 [23] used for M. thermophila transformation, was grown in Luria–Bertani medium at 28 °C and kanamycin was added when needed to maintain binary plasmids [39].
Preparation and transformation of M. thermophila ATCC 42464 mediated by Agrobacterium
M. thermophila transformation mediated by A. tumefaciens was performed using a modification of the method described previously for Fusarium circinatum [21]. A single colony of A. tumefaciens harboring binary vector was inoculated into LB medium supplemented with 50 μg/mL kanamycin and cultivated with shaking of 250 rpm at 28 °C for 12–16 h to an optical density of 0.5–1.0 at 600 nm. Subsequently, cells were harvested, washed twice with induction medium (IM) containing AS and then diluted to an OD600 of 0.15. The culture was grown at 28 °C with shaking at 250 rpm for 6–8 h to an OD600 of 0.5–0.8. Conidia were harvested into 0.05 % Tween 80, filtered through glass wool to remove residual mycelia and adjusted to 106 conidia per milliliter calculated using a hemocytometer. An aliquot of 100 μL A. tumefaciens culture was mixed with an equal volume of M. thermophila conidia and spread on an IM agar plate (9.0 cm) containing AS and was covered with cellophane. After co-cultivation at 28 °C, the cellophane was transferred onto MM medium plate supplemented with 100 μg/mL phosphinothricin (Sigma-Aldrich, St. Louis, USA) and 300 μg/mL cefotaxime (Sigma-Aldrich), overlaid with Vogel’s salt agar medium containing 2 % sucrose and 100 μg/mL phosphinothricin and then cultivated at 45 °C. Putative transformants were visible after approximately 3 days.
Mitotic stability analysis
Enhanced GFP (EGFP) was used as the reporter gene to elucidate mitotic stability of transformants. Twenty randomly selected successful transformants with pPK2BarGFPD were consecutively sub-cultured on agar plates without the selective agent. When the agar plates were full of mycelia and conidia, conidia were used to inoculate the next generation culture. After five generations, the GFP-signal was detected under fluorescence microscopy and PCR analysis targeting the bar gene with the specific primer sets, bar-F/bar-R (Table 1), was performed. Triplicate reactions were performed for each transformant.
Binary vector construction for ku70 disruption
The master Ti vector pPK2BarGFPD [23] containing the phosphinothricin resistant bar marker, was employed to test transformation efficiency of M. thermophila mediated by A. tumefaciens. To disrupt the ku70 gene involved in the nonhomologous end-joining (NHEJ) pathway, the plasmid pPK2-ku70 was constructed (Fig. 3). The 5′ and 3′ flanking fragments (2095 bp and 1246 bp) were amplified from M. thermophila genomic DNA with paired primers (Table 1) designed using the annotated genome of M. thermophila ATCC 42464 [12]. The two flanks were inserted between the XbaI/EcoRI and SpeI/EcoRV sites of pPK2BarGFPD, respectively, to result in pPK2-ku70. The inserted fragments were sequenced to confirm the authenticity of the modified plasmids. All restriction enzymes were obtained from Thermo Scientific (Waltham, MA, USA).
Identification of transformants by PCR and Southern blotting
Transformants harboring pPK2BarGFPD were verified by PCR analysis with paired primers, bar-F/bar-R (Table 1), to detect bar after fluorescence detection under the microscope.
The ku70 deletion mutant transformants with ectopic plasmid insertions were recognized by checking fluorescence under the microscope. The green fluorescent signal suggested it was an ectopic insertion mutant rather than homologous recombination replacement. Proper gene replacement was confirmed by amplifying specific fragments with one primer located in the bar gene and the other located in the downstream of 3′ flank in the genomic DNA (Table 1). Mutants with correct placement of the bar gene underwent further homokaryon analysis. The knockout mutants containing no wild-type open reading fragment of ku70 were determined by PCR analysis using paired primers, ku70-F/ku70-R (Table 1).
To verify the authenticity of ku70 mutants, Southern blotting was performed with 20 μg genomic DNA digested by XhoI. Genomic DNA was extracted as described previously [40]. The digested DNA was separated by agarose gel electrophoresis and DNA transfer was performed as described previously [41]. A 441 bp PCR-amplified product used as the probe was generated with paired primers, Probe-F/Probe-R (Table 1). Probe preparation, membrane hybridization and visualization were performed according to the manufacturer’s instructions (DIG High Prime DNA Labeling and Detection Starter Kit II, Roche, Mannheim, Germany).
pyrG disription in ku70 mutant
The pyrG gene (MYCTH_2311494) encoding orotidine-5'-phosphate decarboxylase was disrupted through homologous replacement in the wild-type strain (WT) and ku70 mutant. The PtrpC and neomycin resistant elements were amplified from pCB1003 and pEGFP-N2 using paired primers PtrpC-F/R and neo-F/R, respectively, and fused using the primers PtrpC-F/neo-R with several restriction sites introduced to both ends. The PtrpC-neo cassette was cloned into the HindIII-BglII sites of pCAMBIA-0380 (CAMBIA, Canberra, Australia; p0380 herein) to generate p0380-neo. The 5′ and 3′ fragments of pyrG (2071 and 1972 bp) were separately amplified from M. thernophila genomic DNA via PCR with paired primers (Table 1). The amplified fragments were inserted into EcoRI/BamHI and XhoI/BglII sites in p0380-neo plasmid, forming the disruption plasmid p0380-pyrGup-neo-pyrGdn. The constructed plasmids were transformed into WT and ku70 deletion strains via Agrobacterium-mediated transformation. Colonies grown for 5 or 6 days on a selective medium at 45 °C were screened in terms of the neomycin resistance to G418 (40 μg mL/L), followed by identification via PCR with paired primers (Table 1).
Microscopy
Confocal microscopy was performed on a Leica TCS SP5 II microscope using a 100× 1.4 NA oil or 20× immersion objective, 30 % Laser (Argon, visible) Power, PMT Trans of 180 HV, Emission bandwidth of PMT was 500 nm to 600 nm. The images were processed and analyzed by ImageJ (version 1.47).
Phenotype characterization of the ku70 mutant
Mature conidia from wild-type and ku70 mutant strains were inoculated into 100 mL 1 × Vogel’s salt supplemented with 1 % Avicel (PH101, Sigma-Aldrich, St. Louis, MO, USA) at 2.5 × 105 conidia per milliliter in a 250-mL Erlenmeyer flask. After growth for 16 h at 45 °C with shaking at 150 rpm under constant light, total secreted protein in the supernatant was determined using a Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA, USA) and 25 μl of supernatant was used for SDS-PAGE analysis. The experiments were performed in biological triplicates.
Abbreviations
- AS:
-
Acetosyringone
- EGFP:
-
Enhanced green fluorescent protein
- DIC:
-
Differential interference contrast
- gDNA:
-
Genomic DNA
- IM:
-
Induction medium
- NHEJ:
-
Nonhomologous end-joining
- PCR:
-
Polymerase chain reaction
- RB and LB:
-
Right and left border of T-DNA, respectively
References
Roy SK, Dey SK, Raha SK, Chakrabarty SL. Purification and properties of an extracellular endoglucanase from Myceliophthora thermophila D-14 (ATCC 48104). J Gen Microbiol. 1990;136(10):1967–71.
Kaur G, Kumar S, Satyanarayana T. Production, characterization and application of a thermostable polygalacturonase of a thermophilic mould Sporotrichum thermophile Apinis. Bioresour Technol. 2004;94(3):239–43.
Vafiadi C, Topakas E, Biely P, Christakopoulos P. Purification, characterization and mass spectrometric sequencing of a thermophilic glucuronoyl esterase from Sporotrichum thermophile. FEMS Microbiol Lett. 2009;296(2):178–84.
Karnaouri A, Topakas E, Antonopoulou I, Christakopoulos P. Genomic insights into the fungal lignocellulolytic system of Myceliophthora thermophila. Front Microbiol. 2014;5:281.
Singh B. Myceliophthora thermophila syn. Sporotrichum thermophile: a thermophilic mould of biotechnological potential. Crit Rev Biotechnol 2014; 15: 1-11.
Gruber F, Visser J, Kubicek CP, Degraaff LH. The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrg-negative mutant strain. Curr Genet. 1990;18(1):71–6.
Punt PJ, van den Hondel CA. Transformation of filamentous fungi based on hygromycin B and phleomycin resistance markers. Methods Enzymol. 1992;216:447–57.
Ruiz-Diez B. Strategies for the transformation of filamentous fungi. J Appl Microbiol. 2002;92(2):189–95.
Visser H, Joosten V, Punt PJ, Gusakov AV, Olson PT, Joosten R, et al. Development of a mature fungal technology and production platform for industrial enzymes based on a Myceliophthora thermophila isolate, previously known as Chrysosporium lucknowense C1. Ind Biotechnol. 2011;7(3):10.
Fungaro MHP, Rech E, Muhlen GS, Vainstein MH, Pascon RC, Dequeiroz MV, et al. Transformation of Aspergillus nidulans by microprojectile bombardment on intact conidia. Fems Microbiol Lett. 1995;125(2-3):293–7.
de Groot MJA, Bundock P, Hooykaas PJJ, Beijersbergen AGM. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol. 1998;16(9):839–42.
Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, et al. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat Biotechnol. 2011;29(10):922–7.
Karnaouri AC, Topakas E, Christakopoulos P. Cloning, expression, and characterization of a thermostable GH7 endoglucanase from Myceliophthora thermophila capable of high-consistency enzymatic liquefaction. Appl Microbiol Biotechnol. 2014;98(1):231–42.
Volkov PV, Rozhkova AM, Gusakov AV, Zorov IN, Sinitsyn AP. Glucoamylases from Penicillium verruculosum and Myceliophthora thermophila: Analysis of differences in activity against polymeric substrates based on 3D model structures of the intact enzymes. Biochimie 2015, 110:45-51.
Topakas E, Moukouli M, Dimarogona M, Christakopoulos P. Expression, characterization and structural modelling of a feruloyl esterase from the thermophilic fungus Myceliophthora thermophila. Appl Microbiol Biotechnol. 2012;94(2):399–411.
Kolbusz MA, Di Falco M, Ishmael N, Marqueteau S, Moisan MC, Baptista Cda S, et al. Transcriptome and exoproteome analysis of utilization of plant-derived biomass by Myceliophthora thermophila. Fungal Genet Biol. 2014;72:10–20.
van den Brink J, van Muiswinkel GC, Theelen B, Hinz SW, de Vries RP. Efficient plant biomass degradation by thermophilic fungus Myceliophthora heterothallica. Appl Environ Microbiol. 2013;79(4):1316–24.
Fang WG, Pei Y, Bidochka MJ. Transformation of Metarhizium anisopliae mediated by Agrobacterium tumefaciens. Can J Microbiol. 2006;52(7):623–6.
Duarte RTD, Staats CC, Fungaro MHP, Schrank A, Vainsten MH, Furlaneto-Maia L, et al. Development of a simple and rapid Agrobacterium tumefaciens-mediated transformation system for the entomopathogenic fungus Metarhizium anisopliae var. acridum. Lett Appl Microbiol. 2007;44(3):248–54.
Gouka RJ, Gerk C, Hooykaas PJJ, Bundock P, Musters W, Verrips CT, et al. Transformation of Aspergillus awamori by Agrobacterium tumefaciens-mediated homologous recombination. Nat Biotechnol. 1999;17(6):598–601.
Covert SF, Kapoor P, Lee M-h, Briley A, Nairn CJ. Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res. 2001;105(3):259–64.
Lin L, Wang F, Wei D. Chlorimuron ethyl as a new selectable marker for disrupting genes in the insect-pathogenic fungus Metarhizium robertsii. J Microbiol Methods. 2011;87(2):241–3.
Lin L, Fang W, Liao X, Wang F, Wei D, St Leger RJ. The MrCYP52 cytochrome P450 monoxygenase gene of Metarhizium robertsii is important for utilizing insect epicuticular hydrocarbons. PLoS One. 2011;6(12):e28984.
Winans SC. Two-way chemical signaling in Agrobacterium-plant interactions. Microbiol Rev. 1992;56(1):12–31.
Bundock P, Dendulkras A, Beijersbergen A, Hooykaas PJJ. Transkingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. Embo J. 1995;14(13):3206–14.
Fang W, Zhang Y, Yang X, Zheng X, Duan H, Li Y, et al. Agrobacterium tumefaciens-mediated transformation of Beauveria bassiana using an herbicide resistance gene as a selection marker. J Invertebr Pathol. 2004;85(1):18–24.
Wang D, He D, Li G, Gao S, Lv H, Shan Q, et al. An efficient tool for random insertional mutagenesis: Agrobacterium tumefaciens-mediated transformation of the filamentous fungus Aspergillus terreus. J Microbiol Methods. 2014;98:114–8.
Shi L, Fang X, Li MJ, Mu DS, Ren A, Tan Q, et al. Development of a simple and efficient transformation system for the basidiomycetous medicinal fungus Ganoderma lucidum. World J Microbiol Biotechnol. 2012;28(1):283–91.
Li M, Zhou L, Liu M, Huang Y, Sun X, Lu F. Construction of an engineering strain producing high yields of alpha-transglucosidase via Agrobacterium tumefaciens-mediated transformation of Asperillus niger. Biosci Biotechnol Biochem. 2013;77(9):1860–6.
Covert SF, Kapoor P, Lee MH, Briley A, Nairn CJ Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res. 2001, 105:259-264
Zhang YJ, Zhao JJ, Xie M, Peng DL. Agrobacterium tumefaciens-mediated transformation in the entomopathogenic fungus Lecanicillium lecanii and development of benzimidazole fungicide resistant strains. J Microbiol Methods. 2014;105:168–73.
Liu Q, Ying SH, Li JG, Tian CG, Feng MG. Insight into the transcriptional regulation of Msn2 required for conidiation, multi-stress responses and virulence of two entomopathogenic fungi. Fungal Genet Biol. 2013;54:42–51.
Yao HZ, Xiao LW, Tian HW, Qiao J. Agrobacterium-mediated transformation (AMT) of Trichoderma reesei as an efficient tool for random insertional mutagenesis. Appl Microbiol Biotechnol. 2007;73(6):1348–54.
Koh CM, Liu Y, Moehninsi, Du M, Ji L. Molecular characterization of KU70 and KU80 homologues and exploitation of a KU70-deficient mutant for improving gene deletion frequency in Rhodosporidium toruloides. BMC Microbiol. 2014;14:50.
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A. 2006;103(27):10352–7.
Guangtao Z, Hartl L, Schuster A, Polak S, Schmoll M, Wang T, et al. Gene targeting in a nonhomologous end joining deficient Hypocrea jecorina. J Biotechnol. 2009;139(2):146–51.
Sorensen LQ, Lysoe E, Larsen JE, Khorsand-Jamal P, Nielsen KF, Frandsen RJ. Genetic transformation of Fusarium avenaceum by Agrobacterium tumefaciens mediated transformation and the development of a USER-Brick vector construction system. BMC Mol Biol. 2014;15:15.
Gauthier GM, Sullivan TD, Gallardo SS, Brandhorst TT, Vanden Wymelenberg AJ, Cuomo CA, et al. SREB, a GATA transcription factor that directs disparate fates in blastomyces dermatitidis including morphogenesis and siderophore biosynthesis. PLoS Pathog. 2010;6(4):e1000846.
Lazo GR, Stein PA, Ludwig RA. A DNA transformation-competent Arabidopsis genomic library in Agrobacterium. Nat biotechnol. 1991;9(10):963–7.
Raeder U, Broda P. Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol. 1985;1(1):17–20.
Sambrook J, Russell DW. Molecular cloning - A laboratory manual, 3rd edition. Cold Spring Harbor Laboratory Press 2001.
Acknowledgements
We thank all members of the Tian laboratory for critical comments and technical assistance, and we thank Lixian Wang from Technology Support Center of Tianjin Institute of Industrial Biotechnology of CAS for her help with the confocal microscopy. This work was supported by the 973 Program of China (2011CBA00800), the 863 Program of China (SS2014AA021300) and Tianjin Zhuanxiang (13ZCDZSY05000).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JX, JL and CT designed the project. JX, LL, QL and WS carried out the experiments. JL, BH and CT wrote the manuscript. All the authors have read the article and approved the final manuscript.
Jing Xu and Jingen Li contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Xu, J., Li, J., Lin, L. et al. Development of genetic tools for Myceliophthora thermophila . BMC Biotechnol 15, 35 (2015). https://doi.org/10.1186/s12896-015-0165-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-015-0165-5