
Abstract
Background
Lower urinary tract symptoms (LUTS) is the most common complication of diabetes. However, the underlying pathogenesis of cultured negative LUTS (cn-LUTS) in diabetic patients has not been well understood. Numerous evidence indicates that urinary dysbiosis is related to urologic disorders. We aim to study alterations of the urinary microbiota of cn-LUTS in type 2 diabetes (T2D) patients.
Methods
Female T2D patients and controls were recruited and requested to finish the American Urological Association Symptom Index. Mid-stream urine was collected for culturing and extracting DNA. Microbial diversity and composition were analyzed by targeting to 16S rDNA. Linear discriminant analysis effect size (LEfSe) was carried out to identify significantly different bacteria.
Results
32 female T2D patients and 26 controls were enrolled. No significant differences in alpha diversity were observed between patients and controls. However, statistically decreased richness (ACE index and Chao 1 index, 85.52(13.75, 204.84) vs. 129.82(63.89, 280.30) and 83.86(11.00, 210.77) vs. 125.19(62.00, 251.77), P = 0.005; Observed Species, 76(10, 175) vs. 98(54, 234), P = 0.011) and decreased species diversity (Shannon index, 1.37(0.04, 3.48) vs. 2.09(0.98, 3.43), P = 0.033; Simpson index, 0.46 (0.06, 0.99) vs. 0.23(0.07, 0.64), P = 0.029) were shown in moderate-to-severe LUTS group and high Hemoglobin A1c group, respectively. A significant difference of beta diversity was found between T2D patients and controls and T2D patients with different severity of cn-LUTS as well as the different level of Hemoglobin A1c. LEfSe revealed that 10 genera (e.g., Escherichia-Shigella and Klebsiella) were increased and 7 genera were decreasing in T2D patients, 3 genera (e.g., Escherichia-Shigella and Campylobacter) were increased and 16 genera (e.g., Prevotella) were reduced in moderate-to-severe LUTS group, 2 genera (Escherichia-Shigella and Lactobacillus) were over-represented and 10 genera (e.g., Prevotella) were under-represented in high Hemoglobin A1c group. Finally, Hemoglobin A1c was found positively correlated with the total score of the American Urological Association Symptom Index (r = 0.509, P = 0.003).
Conclusions
Urinary dysbiosis may be related to cn-LUTS in female T2D patients. A better understanding of urinary microbiota in the development and progression of cn-LUTS in female T2D patients was necessary. The severity of cn-LUTS was correlated to hyperglycemia and chronic hyperglycemia might induce or promote cn-LUTS by influencing urinary microbiota.
Similar content being viewed by others
Background
Diabetes is burgeoning worldwide due to an increase in aging population and changes in eating habits [1]. Chronic hyperglycemia is associated with damage, dysfunction, and failure of multiple organ systems, including the genitourinary system. 80% of individuals with diabetes are accompanied by lower urinary tract symptoms (LUTS) [1, 2]. Although diabetes-induced LUTS is not life-threatening, it seriously affects patients’ life quality. Most diabetic patients are likely suffering from LUTS without evidence of urinary tract infections (UTI), which are refractory and hardly benefit from the conventional treatment. For these patients, it is supposed that the potential pathogenesis of LUTS may be linked to polyuria, oxidative stress and autonomic neuropathy induced by chronic hyperglycemia [3]. However, the underlying pathogenesis of cultured negative LUTS (cn-LUTS) induced by diabetes has not been fully appreciated.
Microbiota has been increasingly considered as an essential factor in human health and disease [4,5,6,7,8]. Traditionally, specimens from mid-stream urine were usually considered being sterile for not enough uropathogens were yielded under standard clinical cultivation procedures. However, the dogma of ‘sterile urine’ has been broken. Recent studies have revealed that urine samples from healthy individuals inhabit numerous microorganisms [9]. Moreover, alterations of urinary microbiota were found in various urologic disorders [10,11,12,13]. Decreasing diversity and richness of urinary microbiota were found in female overactive bladder patients in our previous study [10]. Pearce et al. found that, compared to healthy controls, the proportion of Gardnerella in urge urinary incontinence patients was increased while the proportion of Lactobacillus was decreasing [11]. Siddiqui et al. reported that the diversity of urinary microbiota in interstitial cystitis patients was lower than that in healthy individuals and the proportion of Lactobacillus was increased [12].
An increased level of urine glucose induced by chronic hyperglycemia changes the microenvironment of the urinary tract which might further change the urinary microbiota. Liu et al. reported that urine specimens collected from female Type 2 diabetes (T2D) patients had a decreasing diversity and richness urinary microbiota [14]. They also found that increased Actinobacteria phylum and decreasing Akkermansia muciniphila was associated with the level of fasting blood glucose levels [14]. Accordingly, we suggested that alterations of urinary microbiota might also take part in the occurrence of cn-LUTS in diabetic patients. The goal of our study was to characterize urinary microbiota in Chinese T2D females by comparing the alpha and beta diversity as well as the specific genus and to explore the potential pathogenesis of diabetes-induced cn-LUTS from the characteristics of urinary microbiota.
Methods
Patient enrollment and urinary samples collection
This study was subject to approval by the Ethical Committee of Southern Medical University. From June 2017 to December 2017, females who diagnosed with T2D and healthy female volunteers, age between 40 and 70, were approached for participation. Participants with medical conditions that could interfere with voiding function such as presence of UTI, known abnormal anatomical abnormalities of urinary tract, hereditary and congenital diseases (spina bifida, spinal meninges, etc.), neurological diseases (Guillain-Barré syndrome, multiple sclerosis, etc.), LUTS caused by drug abuse, history of spinal injury, history of hysterectomy, previous major pelvis surgery and evidence of vaginal prolapse were excluded. Patients during the menstrual period or with medical conditions which may interfere with urinary microbiota such as recent antibiotics usage or indwelling catheter (within one month) were also excluded. All participants were requested to offer demographic information and complete the AUA-SI to evaluate the severity of LUTS. Before evaluation, all participants were briefed on the procedure and asked to write informed consent. Mid-stream urine (50 ml) was collected with the labial separation which was supervised by the author (Weina Huang) and 20 ml was left for standard cultivation to exclude UTI. Within 1 h, the rest specimens were immediately shifted to the laboratory and centrifuged at 16,000 g for 10 min. Pellets were kept at − 80 °C until further processing.
DNA extraction, PCR, and MiSeq sequencing
DNA extraction was carried out using the cultured cells protocol supplied with the DNeasy Blood and Tissue Kit (Qiagen, Germany) in a laminar flow hood to avert contamination. The concentration of extracted DNA was tested by the Nanodrop ND-1000 spectrophotometer (Thermo Electron Corporation, USA). Specific primer sets for V3-V4 regions were chosen to perform PCR amplification of 16S rDNA. To assess the contribution of extraneous DNA from reagents, extraction negative controls (no urine) and PCR negative controls (no template) were included as the blank controls. Qiaquick PCR purification kit (Qiagen, Valencia, CA) was selected for purifying the final PCR products from unincorporated nucleotides and primers. Purified samples were normalized to equal DNA concentration and sequenced by Illumina Miseq sequencer (Illumina, Inc., USA).
Statistical analysis
Differences in the demographic and clinical characteristics between groups were tested by Student’s t-test or Mann-Whitney U test (for continuous variables) as well as Pearson’s chi-square test or Fisher’s exact test (for count data). Pearson’s correlation was implemented to detect the relations between clinical data and indices of bacterial bioinformatics. All data were analyzed using SPSS (version 22). All tests were two-sided and P-values < 0.05 was considered statistically significant. PASS programme (PASS 11, NCSS, Kaysville UT, USA) provides estimates of power by simulation.
Bioinformatics analysis
Quantitative Insights Into Microbial Ecology (QIIME, version 1.80) was applied to create an operational taxonomic units (OTUs) table at a default similarity level of 97% based on the 16S rDNA sequence data [15, 16]. Subsequently, chimera detection was performed through the UCHIME method [17]. The representative sequence of each OTU was aligned to Silva and Greengenes database using Ribosomal Database Project Classifier [18].
Five alpha diversity was calculated by mothur software (version 1.31.2) indexes in QIIME including the Observed Species, Chao 1 index, ACE index, Shannon index, and Simpson index. The difference of alpha diversity was evaluated by Mann-Whitney U test in R (version 3.03). Observed Species, Chao1 index, and ACE index represented bacterial richness, while Shannon index and Simpson index were quantitative measures of bacterial diversity that reflecting both species richness and evenness.
To compare microbial composition between groups, principal coordinate analysis (PCoA) was applied on Bray Curtis, weighted UniFrac and unweighted UniFrac distance metrics to generate three-dimensional plots in QIIME. Multiple Response Permutation Procedure (MRPP, a nonparametric test which applied to test beta diversity values between groups) was performed to test the differences of Bray Curtis, weighted UniFrac, unweighted UniFrac distance metrics between groups in QIIME.
Significant bacteria on the relative abundance was tested by Metastats Test. Initial p-value was corrected with the method of Benjamini-Chochberg in R [19]. False discovery rate (FDR) represented the corrected p-value and FDR < 0.05 was considered significant. To identify significantly different bacteria as biomarkers between groups, taxa summaries were reformatted and inputted into Linear discriminant analysis effect size (LEfSe) via the Huttenhower Lab Galaxy Server [20]. Firstly, the Kruskal-Wallis rank sum test and Mann-Whitney U test were applied to identify the significantly different bacteria. Subsequently, linear discriminant analysis (LDA) was used to score the effect size of the different bacteria. Only taxa with logarithmic LDA score greater than 2 were considered significantly enriched.
Results
Demographic and clinical characteristics of participants
Sixty five female volunteers were enrolled, including 35 T2D patients and 30 healthy controls. All the urine samples from these participants were verified cultured negative during the standard culture procedure. However, 3 samples from patients and 4 from controls were excluded due to the sequencing reads could not be achieved. With 32 patients in diabetes group and 26 subjects in the control group (α = 0.05; β = 0.2), we would have 95% power to detect differences at the 0.05 significance level (alpha) using a two-sided Mann-Whitney U test and have 96% power to detect differences at the 0.05 significance level (alpha) using a two-sided Student’s t-test. Detailed comparisons between T2D patients and healthy controls were shown in Table 1. Except for fasting blood glucose, no significant differences were observed in the demographic and clinical characteristics between diabetes group and control group (for example, age, 56.969 ± 8.014 vs. 57.615 ± 9.239; body mass index, 23.739 ± 4.379 vs. 24.298 ± 3.120; hypertension rate, 14(43.8%) vs. 8(30.8%); etc.). Besides, higher scores of American Urological Association Symptom Index (AUA-SI) (total score, 9.219 ± 6.904 vs. 2.846 ± 3.319; storage score, 5.375 ± 4.612 vs. 1.846 ± 1.617; emptying score, 3.844 ± 4.573 vs. 1.000 ± 2.482) were found in T2D patients (P < 0.05).
Comparisons of bioinformatics between T2D patients and controls
All reads were classified into 6212 operational taxonomic units (OTUs). 3070 OTUs were identified in T2D patients and 3142 OTUs were identified in controls. Comparisons of alpha diversity between T2D patients and controls were shown in Table 1. There were no significant differences in Observed Species (88(11, 245) vs. 135(15, 258)), Chao 1 index (103.60(14.00, 272.00) vs. 149.50(29.00, 339.00)), ACE index 104.30(16.50, 416.70) vs. 146.10(40.70, 428.60), Shannon index (1.70(0.04, 3.48) vs. 1.78(0.01, 3.54)) and Simpson index (0.31(0.06, 0.99) vs. 0.38(0.07,1.00)) between diabetes group and control group (Table 1, Fig. 1a, e), which indicated that neither bacterial richness nor species diversity was not significantly changed in the T2D cohort.

Alpha diversity and principal coordinate analysis for the control group and diabetes group urinary microbiota. Observed Species (a); Chao1 index(b); ACE index (c); Shannon index (d); Simpson index (e). Principal coordinate analysis plots of the urinary microbiota based on the weighted UniFrac (f), unweighted UniFrac (g) and Bray-Curtis (h) distance metrics. Association of specific microbiota taxa with the diabetes group and control group was analyzed by LEfSe (i). Genera enriched for controls in red and enriched for patients in blue. Only genera meeting a linear discriminant analysis score threshold > 2 are shown
As shown in Fig. 1f – h, we found urinary microbiota in diabetes group and control group mainly clustered together in the different area of the PCoA plots, respectively. Statistical differences were noted in weighted UniFrac (P = 0.045) as well as unweighted UniFrac (P = 0.010) distance metrics. However, the difference in Bray Curtis distance metrics could not reach statistical significance (Fig. 1h, P = 0.053).
We found that 10 genera were over-represented in the T2D cohort including Escherichia-Shigella, Klebsiella, Aerococcus, Delftia, Enterococcus, Alistipes, Stenotrophomonas, Micrococcus, Deinococcus and Rubellimicrobium, while 7 genera were under-represented including Gallicola, Arcobacter, Arcanobacterium, Kocuria, Murdochiella, Solitalea and Peptoniphilus (Fig. 1i). Among the genera mentioned above, Metastats algorithm shown the significantly altered in Delftia, Enterococcus, Stenotrophomonas, Micrococcus, Deinococcus, Solitalea and Peptoniphilus (Additional file 1: Table S1).
Comparisons of bioinformatics between T2D patients with no to mild LUTS and those with moderate to severe LUTS
In order to detect the specific bacteria associated with the severity of LUTS, we next studied whether the urinary microbial profile was different between T2D patients with no to mild LUTS (LS group, total AUA-SI score ≤ 7) and T2D patients with moderate to severe LUTS (HS group, total AUA-SI score > 7). Higher Observed Species (Fig. 2a, 76(10, 175) vs. 98(54, 234), P = 0.011), Chao1 index (Fig. 2b, 83.86(11.00, 210.77) vs. 125.19(62.00, 251.77), P = 0.005), ACE index (Fig. 2c, 85.52(13.75, 204.84) vs. 129.82(63.89, 280.30), P = 0.005) were presented in the LS group, while no significant differences were observed in Shannon index (Fig. 2d, 1.40(0.04, 3.48) vs. 2.01(0.98, 3.43), P = 0.132) and Simpson index (Fig. 2e, 0.44(0.06, 0.99) vs. 0.28(0.07, 0.64), P = 0.202). This indicated that significantly decreasing bacterial richness was found in the HS group (Table 2). However, no correlation was observed between AUA-SI and alpha diversity.

Alpha diversity and principal coordinate analysis for the HS group and LS group urinary microbiota. Observed Species (a); Chao1 index (b); ACE index (c); Shannon index (d); Simpson index (e). Principal coordinate analysis plots of the urinary microbiota based on the weighted UniFrac (f), unweighted UniFrac (g) and Bray-Curtis (h) distance metrics. Association of specific microbiota taxa with the HS group and LS group was analyzed by LEfSe (i). Genera enriched for HS in red and enriched for LS in blue. Only genera meeting a linear discriminant analysis score threshold > 2 are shown
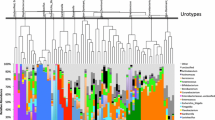
PCoA plots demonstrated that samples from different groups were clustered together respectively and suggested that the urinary microbiota composition of LS group was distinct from that of HS group (Fig. 2f, h). Statistically differences were observed in MRPP test (P = 0.002, P = 0.024, P = 0.001 for weighted UniFrac, unweighted UniFrac and Bray Curtis distance metrics, respectively). In addition, a clear hierarchical clustering of LS samples and HS samples was observed on dendrogram which based on Bray Curtis distance metrics (Fig. 3).

A clear hierarchical clustering of HS samples was observed in the dendrogram at the genus level (left; based on Bray Curtis distance metrics). In the histogram (right), each colored box represents a bacterial taxon and each bar, a subject. The height of a colored box represents the relative abundance of each organism in the sample. Bacterial genera with a relative abundance < 0.5% and unclassified genera are grouped as “Other”
LEfSe revealed that 3 genera were over-represented in the HS group including Escherichia-Shigella, Campylobacter, Megasphaera, while 16 genera were under-represented including Prevotella, Dialister, Leptotrichia, Anaerococcus, Fusobacterium, Prevotella_6, Fastidiosipila, Varibaculum, Mycoplasma, Peptoniphilus, Porphyromonas, Fodinicola, Brevundimonas, Candidatus-Nomurabacteria, Paracoccus and Rhodococcus (Fig. 2i). The Metastats analysis shown that Escherichia-Shigella was significantly more abundant in the HS group, while Prevotella, Leptotrichia, Varibaculum, Mycoplasma, and Candidatus-Nomurabacteria were more abundant in the LS group (Additional file 1: Table S1).
Of interest was that no significant differences were noted in clinical characteristics except Hemoglobin A1c (HbA1c) between the LS group and HS group (Table 2).
Comparisons of bioinformatics between T2D patients with low HbA1c and high HBA1c
Due to the significant difference of HbA1c was found between LS group and HS group, we further analysis the characteristics of urinary microbiota between female T2D patients with low HbA1c (LH group, HbA1c ≤ 7%) and high HbA1c (HH group, HbA1c > 7%). Higher Shannon index (Fig. 4d, 1.369(0.044, 3.478) vs. 2.089(0.979, 3.429), P = 0.033) and lower Simpson index (Fig. 4e, 0.461(0.055, 0.987) vs. 0.229(0.069, 0.640), P = 0.029) were presented in LH group, while no significantly difference in Observed Species, Chao1 index and ACE index (Fig. 4a, 77(10,204) vs. 86(54,234), P = 0.157; Fig. 4b, 97.00(11.00, 213.55) vs. 115.67(63.89,245.84), P = 0.105; Fig. 4c, 94.87(0.26, 3.32) vs. 121.30(63.89, 280.30), P = 0.089). Comparison of alpha diversity indicated that significantly decreasing species diversity was discovered in HH cohort.

Alpha diversity and principal coordinate analysis for HH group and LH group urinary microbiota. Observed Species (a); Chao1 index (b); ACE index (c); Shannon index (d); Simpson index (e). Principal coordinate analysis plots of the urinary microbiota based on the weighted UniFrac (f), unweighted UniFrac (g) and Bray-Curtis (h) distance metrics. Association of specific microbiota taxa with HH group and LH group was analyzed by LEfSe (i). Genera enriched for HH in red and enriched for LH in blue. Only genera meeting a linear discriminant analysis score threshold > 2 are shown
PCoA plots demonstrated that LH group samples and HH group samples were clustered respectively, which suggested that the urinary microbiota composition of LH group differed from that of HH group (Fig. 4f and Fig. 4h). MRPP test shown that the observed differences in PCoA plots were statistically significant (P = 0.001 for weighted UniFrac and Bray Curtis distance metrics, respectively). However, the difference in unweighted UniFrac distance metrics could not reach statistical significance (Fig. 4g, P = 0.078).
LEfSe demonstrated that 2 genera were over-represented in the HH group, including Escherichia-Shigella and Lactobacillus, while 10 genera were over-represented in the LH group, including Prevotella, Campylobacter, Dialister, Anaerococcus, Peptoniphilus, Porphyromonas, Fodinicola, Negativicoccus (Fig. 4i). Among these genera, Metastats algorithm shown the significantly different in Anaerococcus, Fodinicola, Lactobacillus, Peptoniphilus, Senegalimassilia (Additional file 1: Table S1).
Higher scores of AUA-SI (total score, 13.059 ± 6.805 vs. 4.867 ± 3.796; storage score, 7.824 ± 4.825 vs. 2.600 ± 2.230; emptying score, 5.235 ± 5.118 vs. 2.267 ± 3.369) were found in HH group (P < 0.05, Additional file 2: Table S2). Besides, HbA1c was found positively correlated with the total score of AUA-SI based on Pearson’s correlation (Fig. 5, r = 0.509, P = 0.003).

Correlations between AUA-SI and Hemoglobin A1c in female type 2 diabetes patients. Higher Hemoglobin A1c positively correlated with the AUA-SI score
Discussion
In this study, we describe the urinary microbiota in the female with and without T2D, female T2D patients with no to mild LUTS and moderate to severe LUTS as well as patients with low HbA1c and high HBA1c using 16S rDNA sequencing. Based on other reports [10, 11, 21] and our data, this is further confirmation that the commonly held clinical belief that healthy urine should be sterile is false.
Consented with Liu’s study, our results indicated that the composition of urinary microbiota may be impacted by diabetes [14]. (1) Samples from T2D patients and those from controls clustered separately in PCoA plots (Fig. 1f, h), which suggested a common urinary dysbiosis was associated to diabetes; (2) 10 genera were over-represented in T2D patients, including common pathogens such as Escherichia-Shigella, Klebsiella and Enterococcus; (3) it is found that female T2D patients suffering from much serious LUTS. Taken together, these results indicated that urinary dysbiosis (e.g. significantly increasing pathogens) may contribute to the initiation or development of cn-LUTS in type 2 diabetic patients. Common pathogens that over-represented in T2D patients would be discussed below.
Urinary microbial profile between LS and HS group was further analyzed. We found that the composition of urinary microbiota in the LS group was also significantly different from that in the HS group (Fig. 2f, 2i, Fig. 3). Hence, this raises the possibility that specific microbial patterns may be associated with more serious LUTS or the progression of LUTS in the female T2D patients. We may find out the specific urinary microbiota (Fig. 2i) that might exacerbate diabetes-induced cn-LUTS. The relatively increasing and decreasing genus would be also discussed in the following passage.
Due to the significant difference of HbA1c was found between the LS group and HS group, urinary microbial characteristics in the HH group and LH group were also analyzed. Statistical differences of urinary microbiota composition between HH and LH cohort were found in our analysis (Fig. 4f, i). In Liu et al.’s study, they found that increased Actinobacteria phylum as well as decreasing Akkermansia muciniphila were positively correlated with fasting blood glucose [14]. It is therefore suggested that poorly controlled blood glucose might further influence the composition of urinary microbiota. Furthermore, we found that most patients in the HH group suffered from more serious LUTS and HbA1c was positively correlated with AUA-SI. These raise the possibility that poorly controlled blood glucose might exacerbate cn-LUTS. As a result, we suggested the intriguing hypothesis that chronic hyperglycemia might induce or deteriorate cn-LUTS in T2D patients through influencing the urinary microbiota composition. Although the cause-effect relationship among hyperglycemia, urinary microbiota and cn-LUTS cannot be determined under our study, it is worth further researching.
In both cohorts, one or two genera dominated most of the sequence profiles. Escherichia-Shigella was relatively increased in diabetes group and HS group. Klebsiella and Enterococcus were relatively increased in the diabetes group. Klebsiella is a common conditional pathogenic bacterium that causing respiratory and urinary tract infections when host immunity is impaired [22]. Enterococcus is a nosocomial pathogen which can cause UTI and endocarditis [23]. Increased TNF-α, a proinflammatory cytokine, was discovered in the bladder tissue of T2D mice [24]. Toll-like receptors 4 pathway was also activated in type 1 diabetes rats induced by Streptozotocin [25]. It was a strong indication that inflammation existed in the bladder tissue of diabetic animals. In the present study, diabetic patients that included were suffered from cn-LUTS. As for these patients, standard clinical cultivation procedures have a too low sensitivity to detect the uropathogens. 16S rDNA sequencing of mid-stream urine samples might represent a novel method for diagnosing “UTI” (urinary dysbiosis).
The trends of Prevotella abundance changes were variable in different researches. In our study, Prevotella was decreasing in HS cohort and our previous study shown that decreasing Prevotella was found in overactive bladder patients [10]. However, Pearce et al. revealed that enrichment of Prevotella was observed in urgency urinary incontinence patients [11]. 16S rDNA sequencing-based approach can’t identify bacteria well at the species level and over 20 species have been already classified into Prevotella. It is hypothesized that various species play distinct roles in different disorders, leading to the contradiction mentioned before. Thus, it may be necessary to explore the role of Prevotella in urologic disorders via whole genome shotgun sequencing or expanded urine culture.
Enrichment of Lactobacillus was presented in the HH group. Lactobacillus is considered being low virulence, hardly causing infections in human beings [12]. It is best known for its dominance in vaginal microbiota and it can prevent vaginitis by maintaining a physiological acidic environment in the vagina. Because of these properties, Lactobacillus has been applied to prevent or even treat UTI [26]. However, there is increasing evidence that specific Lactobacillus spp may be pathogenic and linked to UTI [27, 28]. It has been suggested that Lactobacillus is resistant to the widely used antibiotics, which can reproduce during the treatment of UTI and invade the bladder to induce inflammation [29]. In addition, several studies found that Lactobacillus was closely associated with cn-LUTS [27, 28]. For example, Maskell et al. found that Lactobacillus gradually disappeared (detected by special culture techniques) as symptoms subsided after the use of antibiotics for treating LUTS in the female patients who had a negative urine culture before [30]. The result suggested that a shift in the microbial community towards Lactobacillus in the HH group may be an important etiological factor for the more serious LUTS reported by the patients.
In this study, no significant differences in alpha diversity were found between T2D patients and controls but decreased richness as well as species diversity were shown in HS and HH group, respectively. Although a reduction in alpha diversity has been thought as a feature of gastrointestinal diseases, such as ulcerative colitis, Crohn’s disease and colorectal cancer [31,32,33] as well as an increased vaginal microbiota diversity was associated with bacterial vaginitis [34]. No consistent changes in alpha diversity were found in urologic disorders. Increased alpha diversity was observed in urgency urinary incontinence [11], decreasing alpha diversity was demonstrated in overactive bladder [10], while no significant difference was noted in prostate cancer [35]. In Liu et al.’s study, they reported that urinary microbiota diversity and richness were lower in female T2D patients [14]. Demographic features, exclusive criteria and less microbial species in the urinary tract may be the reasons leading to this contradiction between Liu’s study and ours [14].
Owing to the urine samples may be contaminated with microbiota surrounding the urethral orifice, mid-stream urine was collected by the clean catch method with labial separation supervised by the author (Weina Huang). Catheter-derived urine samples were suggested as an alternative method, but the method was considered being invasive and was not ethically workable in patients who do not have a clinical indication for it. Comparison of results from Siddiqui et al.’s urinary microbiota study on female mid-stream urine with the consequences of suprapubic aspirate by Wolfe et al. shown that the major findings were the same [12]. It was a strong indication that results from mid-stream urinary microbiota were reliable.
Participants who used antibiotics recently have been excluded, however, numerous drugs may influence gut microbiota composition, and their effect may be present even in the urinary microbiota. Anti-diabetic drugs, particularly metformin, also have a well-known effect on the gut microbiota composition [36]. Although all the T2D patients included in our study were hospitalized and most of them were treated with insulin (data not shown), we could not fully avoid the interference of anti-diabetic drugs. The correlation between urinary microbiota and anti-diabetic drugs should be studied.
Our study is not devoid of limitations. Firstly, it is impossible to determine the cause-effect relationship between symptoms and bioinformatics indicators in this cross-sectional study. Thus, prospective studies with a larger sample and animal experimentation will be needed to clarify the role of urinary microbiota in the development and progression of cn-LUTS in female T2D patients. Secondly, we cannot confirm that the urinary microbes characterized by 16S rDNA sequencing were actually viable. We should add expanded urine culture as a complementary method and make a comprehensive analysis of information that provided by expanded urine culture and 16S sequencing in the future. Finally, the heterogeneity of our cohort should not be ignored because of wide the range of age and duration of diabetes in our cohort. Thus, patients might be stratified by age and time of symptoms in a larger cohort.
Conclusions
We have comprehensively profiled the urinary microbiota associated with cn-LUTS in female T2D patients. The link between urinary microbiota and cn-LUTS in female T2D patients without evidence of UTI has been established but the cause-effect relationship is still unclear. A better understanding of urinary microbiota in the development and progression of cn-LUTS in female T2D patients was necessary, which might provide novel diagnostic biomarkers as well as microbiota-targeted therapeutic options. The severity of cn-LUTS might be related to hyperglycemia and chronic hyperglycemia might induce or promote cn-LUTS by influencing the composition of urinary microbiota.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AUA-SI:
-
American Urological Association Symptom Index questionnaire
- cn-LUTS:
-
Cultured negative lower urinary tract symptoms
- HbA1c:
-
Hemoglobin A1c
- LEfSe:
-
Linear discriminant analysis effect size
- LUTS:
-
Lower urinary tract symptoms
- MRPP:
-
Multiple Response Permutation Procedure
- OTUs:
-
Operational taxonomic units
- PCoA:
-
Principal coordinate analysis
- QIIME:
-
Quantitative Insights Into Microbial Ecology
- T2D:
-
Type 2 diabetes
- UTI:
-
Urinary tract infections
References
Kirschner-Hermanns R, Daneshgari F, Vahabi B, Birder L, Oelke M, Chacko S. Does diabetes mellitus-induced bladder remodeling affect lower urinary tract function? ICI-RS 2011. Neurourol Urodyn. 2012;31:359–64.
Golbidi S, Laher I. Bladder dysfunction in diabetes mellitus. Front Pharmacol. 2010;1:136.
Daneshgari F, Liu G, Birder L, Hanna-Mitchell AT, Chacko S. Diabetic bladder dysfunction: current translational knowledge. J Urol. 2009;182(Suppl):18–26.
Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ. 2017;15:j831.
Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 2010;8:481–90.
Irrazábal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014;54:309–20.
Chu H, Khosravi A, Kusumawardhani IP, Kwon AH, Vasconcelos AC, Cunha LD, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science. 2016;352:1116–20.
Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with breast Cancer. Appl Environ Microbiol. 2016;82:5039–48.
Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, Fitzgerald M, et al. Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol. 2012;50:1376–83.
Wu P, Chen Y, Zhao J, Zhang G, Chen J, Wang J, et al. Urinary microbiome and psychological factors in women with overactive bladder. Front Cell Infect Microbiol. 2017;7:488.
Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, et al. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. MBio. 2014;5:e01283–14.
Siddiqui H, Lagesen K, Nederbragt AJ, Jeansson SL, Jakobsen KS. Alterations of microbiota in urine from women with interstitial cystitis. BMC Microbiol. 2012;12:205.
Fouts DE, Pieper R, Szpakowski S, Pohl H, Knoblach S, Suh MJ, et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Transl Med. 2012;10:174.
Liu F, Ling Z, Xiao Y, Lv L, Yang Q, Wang B, et al. Dysbiosis of urinary microbiota is positively correlated with type 2 diabetes mellitus. Oncotarget. 2017;8:3798–810.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Wu P, Zhang G, Zhao J, Chen J, Chen Y, Huang W, et al. Profiling the urinary microbiota in male patients with bladder Cancer in China. Front Cell Infect Microbiol. 2018;8:167.
Dehshiri M, Khoramrooz SS, Zoladl M, Khosravani SA, Parhizgari N, Motazedian MH, et al. The frequency of Klebsiella pneumonia encoding genes for CTX-M, TEM-1 and SHV-1 extended-spectrum beta lactamases enzymes isolated from urinary tract infection. Ann Clin Microbiol Antimicrob. 2018;17(1):4.
Fisher K, Phillips C. The ecology, epidemiology and virulence of Enterococcus. Microbiology. 2009;155:1749–57.
Wang Z, Cheng Z, Cristofaro V, Li J, Xiao X, Gomez P, et al. Inhibition of TNF-α improves the bladder dysfunction that is associated with type 2 diabetes. Diabetes. 2012;61:2134–45.
Szasz T, Wenceslau CF, Burgess B, Nunes KP, Webb RC. Toll-like receptor 4 activation contributes to diabetic bladder dysfunction in a murine model of type 1 diabetes. Diabetes. 2016;65:3754–64.
Falagas ME, Betsi GI, Tokas T, Athanasiou S. Probiotics for prevention of recurrent urinary tract infections in women: a review of the evidence from microbiological and clinical studies. Drugs. 2006;66:1253–61.
Darbro BW, Petroelje BK, Doern GV. Lactobacillus delbrueckii as the cause of urinary tract infection. J Clin Microbiol. 2009;47:275–7.
Imirzalioglu C, Hain T, Chakraborty T, Domann E. Hidden pathogens uncovered: metagenomic analysis of urinary tract infections. Andrologia. 2008;40:66–71.
Maskell RM. The natural history of urinary tract infection in women. Med Hypotheses. 2010;74:802–6.
Maskell R, Pead L, Sanderson RA. Fastidious bacteria and the urethral syndrome: a 2-year clinical and bacteriological study of 51 women. Lancet. 1983;2:1277–80.
Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105:1907–11.
Lepage P, Häsler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141:227–36.
Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–92.
Liu MB, Xu SR, He Y, Deng GH, Sheng HF, Huang XM, et al. Diverse vaginal microbiomes in reproductive-age women with vulvovaginal candidiasis. PLoS One. 2013;8:e79812.
Shrestha E, White JR, Yu SH, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate Cancer. J Urol. 2018;199:161–71.
Tong X, Xu J, Lian F, Yu X, Zhao Y, Xu L, et al. Structural alteration of gut microbiota during the amelioration of human type 2 diabetes with hyperlipidemia by metformin and a traditional Chinese herbal formula: a multicenter, randomized. Open Label Clinical Trial MBio. 2018;9:e02392–17.
Acknowledgments
Not applicable.
Funding
The current work was supported partly by the Natural Science Foundation of Guangdong Province (2018A030313148 for purchasing most of the experimental reagents, materials and sequencing fees) and partly by the National Natural Science Foundation of China (81870522 for providing the labour costs for the researchers and the fee for some antibodies). The funding body support our job in the design of the study.
Author information
Authors and Affiliations
Contributions
PW (Peng Wu), JC (Jiawei Chen) and JZ1 (Jie Zhao) were involved in study design; JC, YC1 (Ying Cao) and GZ (Guihao Zhang) processed the samples and carried out the molecular techniques; JC, JZ2 (Jialei Zhong) and YC2 (Yang Chen) performed the bioinformatics and taxonomic analysis; PW, JC and JZ1 drafted the manuscript; PW, GZ and WH (Weina Huang) performed the critical revision of the manuscript; JZ3 (Jiarong Zeng) performed the statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethical Committee of Southern Medical University; all participants were briefed on the procedure and asked to write informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Differences of urinary microbiota between groups at genus level based on Metastats algorithm (XLSX 12 kb)
Additional file 2:
Table S2. Comparisons of demographic and clinical characteristics between HH group and LH group (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, J., Zhao, J., Cao, Y. et al. Relationship between alterations of urinary microbiota and cultured negative lower urinary tract symptoms in female type 2 diabetes patients. BMC Urol 19, 78 (2019). https://doi.org/10.1186/s12894-019-0506-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-019-0506-0