
Abstract
Background
Members of the Ewing’s sarcoma family of tumor (ESFT) are malignant neoplasms and rarely observed in the adrenal gland.
Case presentation
We report an extremely exceptional case of ESFT rising from the adrenal gland in a 57-year-old Chinese man. The patient was hospitalized with abdominal swelling for 2 months. Computed tomography (CT) scan revealed a nearly-circular mass measuring about 8.1 × 10.6 cm in the right adrenal region. The patient underwent right adrenal resection. Histopathologic examination found the tumor was composed of small round blue cells forming typical Homer-Wright rosettes in focal area. The immunohistochemical analysis confirmed the case to be ESFT, which was positive for membranous CD99 and nuclear FLI-1. The patient was scheduled for four courses of large doses of chemotherapy and died for cancer metastasis one year later after surgery.
Conclusions
Histopathological evidence of Homer-Wright rosettes and immunohistochemical markers positivity, such as CD99 and FLI-1, are valuable factors for ESFT diagnosis, although cytogenetic analysis is considered as the gold standard. Complete surgery is the treatment of choice for ESFT and adjuvant radiotherapy and combination chemotherapy can significantly improve the survival rate of postoperative patients.
Similar content being viewed by others


Background
The Ewing’s sarcoma family of tumor (ESFT) are rare aggressive malignancies and consist of Ewing’s sarcoma (ES) of bone, extraosseous Ewing’s, primitive neuroectodermal tumor (PNET), and Askin’s tumor [1, 2]. These distinct entities are characterized by common histopathological and immunohistochemical features, including a primitive undifferentiated small round blue cell associated to a variable level of palisading and rosette formation, as well as strongly positive for the cell surface glycoprotein CD99 [3–5]. The defining feature of the ESFT is a nonrandom chromosomal translocation and the most frequent is EWS-FLI1 fusion [6, 7]. These highly aggressive malignancies most commonly arise in the soft tissue or bone in adolescents and young adults [8]. Reports of cases arising from the adrenal gland are extremely rare. To the best of our knowledge, there are 32 cases in the English literatures [5, 9–30]. We report an additional ESFT case arising from the adrenal gland and discuss its clinical and histopathological characteristics, as well as unusual therapeutic strategies.
Case presentation
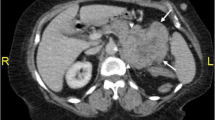
A 57-year-old man presented to the First Hospital of Jilin University (Changchun, China) with the main complaint of abdominal swelling for 2 months. In addition to the mild percussion pain in the right kidney region, no other symptoms were noted during a physical examination. His past medical history was unremarkable. Computed tomography (CT) scan of the abdomen revealed a nearly-circular mass measuring about 8.1 × 10.6 cm arising from the right adrenal gland (Fig. 1a). The CT also showed heterogeneous density, both solid and cystic components and calcification of the mass. The lesion showed heterogeneous enhancement and relatively sharp margination on Contrast-enhanced CT (Fig. 1b). Contrast-enhanced CT scan further defined the large mass was located between the liver and kidney with characteristics consistent with the soft tissue. Vena cava, right renal vein were compressed and displaced. No obvious metastasis was apparent.

Abdominal computed tomography (CT) scan revealed a large mass (arrow) arising from the right adrenal gland (a). The lesion showed heterogeneous enhancement and relatively sharp margination (arrow) on Contrast-enhanced CT (b)
The patient underwent open surgery under general anesthesia. A 10.0 cm × 8.0 cm × 6.0 cm mass was found during laparotomy. The tumor was located above the left renal vein and the right renal vein without venous involvement. Due to firmly adhesion with the surrounding tissue, tumor dissection was difficult. Intraoperative blood loss was 800 mL and the tumor was completely removed eventually. Postoperative histopathology showed a monotonous population of small round blue cells with occasional Homer-Wright-type rosettes (Fig. 2). The results confirmed the diagnosis of PNET. The immunohistochemical staining was performed supporting the previous diagnosis, which was positive for CD99, FLI-1, NeuN, CGA and VIMENTIN (Fig. 2), while negative for EMA, SYN and LCA.

Histopathologic examination showed small round blue cells forming Homer-Wright-type rosettes (H&E, ×400). Immunohistochemical staining revealed the tumor cells were positive for CD 99, FLI-1, NeuN, CGA and VIMENTIN (original magnification × 400)
The patient was scheduled for adjuvant chemotherapy with adriamycin, cyclophosphamide, ifosfamide and etoposide. At his follow-up, 5 months after surgery, CT scan results demonstrated a metastatic lesion arising from the right abdominal wall. Unfortunately, the patient died for cancer metastasis one year later after surgery.
Discussion
ESFT rising from the adrenal gland is extremely exceptional but malignant. Patients often present with tumor compression, flank pain or mass. However, its preoperative imaging diagnosis is difficult and histopathological and genetic tools are required for an accurate diagnosis.
Histopathologically, ESFT appear as immature or primitive small round blue cell tumors infiltrating the soft tissue or bone in a diffuse or lobular pattern. The tumor cells have round to oval nuclei with coarsely stippled chromatin and indistinct nucleoli. The scanty cytoplasm is pale or clear. In addition, these cells are often accompanied by hemorrhage and necrosis. ESFT are mainly represented by the existence of typical Homer-Wright-type rosette or other types of rosettes [17, 31].
Immunohistochemical markers such as CD99, FLI-1, HNK-1 and CAV-1 are commonly expressed in ESFT and provide valuable support to the definitive diagnosis. CD99, a 32-kDa cell surface glycoprotein, is encoded by the MIC2 gene and extremely sensitive for ESFT [4, 14]. The sensitivity is as high as 95% although the specificity is low [14, 31]. Its expression is also observed in T-lymphoblastic lymphoma, rhabdomyosarcoma, synovial sarcoma, and small cell anaplastic osteosarcoma [32–35]. ESFT can be potentially misdiagnosed based merely on expression of CD99. Even so, CD99 is still the most reliable immunohistochemical marker for ESFT. FLI-1, as well as HNK-1, appears reliable but less sensitive for ESFT than CD99 [4, 31]. All authors agree that both markers are expressed in various other round cell tumors [36]. CD99 and FLI-1 are mainly used for the diagnosis of ESFT and an immunohistochemical panel consisting at least these two makers is recommended [37–39]. CAV1, a membrane protein, its high expression is associated with the anchorage-independent growth [40, 41]. Express CAV1 have been shown to be more aggressive and metastatic [41]. CAV1 appears as a diagnostic immunohistochemical marker of ESFT being positive in CD99-negative cases [31]. In addition, markers of NSE, VIMENTIN, cytokeratin and S-100 have been detected in a subset of ESFT by immunohistochemistry.
At present, cytogenetic analysis is the “gold standard” for diagnosis of ESFT. Conventional tests are valuable to make the definitive diagnosis such as Southern blot, Northern blot analyses, FISH and RT-PCR [14, 39, 42]. The diagnosis of our case, ESFT rising from the adrenal gland, was not based on the cytogenetic findings. However, it was supported by the histopathological findings of poorly differentiated, small round blue cells forming typical Homer-Wright rosettes and the immunohistochemical findings of strongly positive for CD99, FLI-1 and negative for differentiation markers such as epithelial sufficiently.
ESFT is an aggressive malignancy with very poor prognosis [6]. Multimodality regimens including surgical resection, adjuvant chemotherapy and radiation therapy are often required [43]. Current surgical approaches include open, laparoscopic and robotic resection. The latter two are more difficult to perform because the large tumor is often accompanied by liquefaction and/or necrosis. Jacob Stephenson et al. [5] reported a ESFT arising from adrenal gland, during operation with the robotic assistance, the tumor capsule was ruptured, which may lead to metastasis and increase the dose of chemotherapy and radiotherapy. Hence, the surgical approach should be selected in accordance with patient’s condition.
Cooperative group studies have led to chemotherapy regimens using the same drugs (vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide), although the exact regimens differ in Europe and North America [2]. Only 16 cases of ESFT arising from the adrenal gland have been reported since 2011. Eleven of these sixteen patients received surgery. Nine received adjuvant chemotherapy and five received radiation treatment. Only two patients with small mass and no evidence of metastasis are alive and disease free. The two long-term survival of patients received multimodality regimens using a combination of complete surgery, as well as chemotherapy and radiotherapy (Table 1). We conclude that complete surgery is the treatment of choice for ESFT. Adjuvant chemotherapy and postoperative radiotherapy have shown significant improvements in survival. The tumor size and metastases are predictors for survival and effect prognosis obviously.
Conclusion
ESFT rising from the adrenal gland is a rare clinical entity. Histopathological evidence of Homer-Wright is crucial for ESFT diagnosis. The neural markers, such as CD99, FLI-1, HNK-1 and CAV-1, may play a valuable role in the immunohistochemical diagnosis of ESFT. The definitive diagnosis of ESFT requires a combination of immunohistochemical examination, as well as histopathologic evaluation, although the “gold standard” will obviously remain cytogenetic analysis. Complete surgery is the treatment of choice for ESFT. Adjuvant chemotherapy and postoperative radiotherapy have shown significant improvements in survival. The tumor size and metastases are predictors for survival and effect prognosis obviously.
Abbreviations
- CAV-1:
-
Caveolin-1
- CD99:
-
Cluster of differentiation 99
- CGA:
-
Chromogranin A
- CT:
-
Computed tomography
- ES:
-
Ewing’s sarcoma
- ESFT:
-
Ewing’s sarcoma family of tumor
- LCA:
-
Leukocyte common antigen
- NSE:
-
Neuron-specific enolase
- PNET:
-
Primitive neuroectodermal tumor
- SYN:
-
Synaptophysin
References
Gupta AA, Pappo A, Saunders N, Hopyan S, Ferguson P, Wunder J, O’Sullivan B, Catton C, Greenberg M, Blackstein M. Clinical Outcome of Children and Adults With Localized Ewing Sarcoma Impact of Chemotherapy Dose and Timing of Local Therapy. Cancer. 2010;116(13):3189–94.
Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncology. 2010;11(2):184–92.
Fagone P, Nicoletti F, Salvatorelli L, Musumeci G, Magro G. Cyclin D1 and Ewing’s sarcoma/PNET: A microarray analysis. Acta Histochem. 2015;117(8):824–8.
Hung YP, Fletcher CDM, Hornick JL. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol. 2016;29(4):370–80.
Stephenson J, Gow KW, Meehan J, Hawkins DS, Avansino J. Ewing sarcoma/primitive neuroectodermal tumor arising from the adrenal gland in an adolescent. Pediatr Blood Cancer. 2011;57(4):691–2.
Tilan JU, Krailo M, Barkauskas DA, Galli S, Mtaweh H, Long J, Wang H, Hawkins K, Lu C, Jeha D, et al. Systemic Levels of Neuropeptide Y and Dipeptidyl Peptidase Activity in Patients With Ewing Sarcoma-Associations With Tumor Phenotype and Survival. Cancer. 2015;121(5):697–707.
Hameiri-Grossman M, Porat-Klein A, Yaniv I, Ash S, Cohen IJ, Kodman Y, Haklai R, Elad-Sfadia G, Kloog Y, Chepurko E, et al. The association between let-7, RAS and HIF-1 alpha in Ewing Sarcoma tumor growth. Oncotarget. 2015;6(32):33834–48.
Lee J, Hoang BH, Ziogas A, Zell JA. Analysis of Prognostic Factors in Ewing Sarcoma Using a Population-Based Cancer Registry. Cancer. 2010;116(8):1964–73.
Marina NM, Etcubanas E, Parham DM, Bowman LC, Green A. Peripheral Primitive Neuroectodermal Tumor (Peripheral Neuroepithelioma) In Children - A Review Of The St Jude Experience And Controversies In Diagnosis And Management. Cancer. 1989;64(9):1952–60.
Renshaw AA, PerezAtayde AR, Fletcher JA, Granter SR. Cytology of typical and atypical Ewing’s sarcoma PNET. Am J Clin Pathol. 1996;106(5):620–4.
Matsuoka Y, Fujii Y, Akashi T, Gosehi N, Kihara K. Primitive neuroectodermal tumour of the adrenal gland. BJU Int. 1999;83(4):515–6.
Pirani JF, Woolums CS, Dishop MK, Herman JR. Primitive neuroectodermal tumor of the adrenal gland. Journal of Urology. 2000;163(6):1855–6.
Kato K, Kato Y, Ijiri R, Misugi K, Nanba I, Nagai J-I, Nagahara N, Kigasawa H, Toyoda Y, Nishi T, et al. Ewing’s sarcoma family of tumor arising in the adrenal gland—Possible diagnostic pitfall in pediatric pathology: Histologic, immunohistochemical, ultrastructural, and molecular study. Hum Pathol. 2001;32(9):1012–6.
Ahmed AA, Nava VE, Pham T, Taubenberger JK, Lichy JH, Sorbara L, Raffeld M, Mackall CL, Tsokos M. Ewing sarcoma family of tumors in unusual sites: Confirmation by RT-PCR. Pediatr Dev Pathol. 2006;9(6):488–95.
Kim MS, Kim B, Park CS, Song SY, Lee EJ, Park NH, Kim HS, Kim SH, Cho KS. Radiologic findings of peripheral primitive neuroectodermal tumor arising in the retroperitoneum. Am J Roentgenol. 2006;186(4):1125–32.
Komatsu S, Watanabe R, Naito M, Mizusawa T, Obara K, Nishiyama T, Takahashi K. Primitive neuroectodermal tumor of the adrenal gland. Int J Urol. 2006;13(5):606–7.
Zhang Y, Li H. Primitive Neuroectodermal Tumors of Adrenal Gland. Jpn J Clin Oncol. 2010;40(8):800–4.
Mohsin R, Hashmi A, Mubarak M, Sultan G, Shehzad A, Qayum A, Naqvi SA, Rizvi SA. Primitive neuroectodermal tumor/Ewing’s sarcoma in adult uro-oncology: A case series from a developing country. Urology annals. 2011;3(2):103–7.
Saboo SS, Krajewski KM, Jagannathan JP, Ramaiya N. IVC tumor thrombus: an advanced case of rare extraosseous Ewing sarcoma of the adrenal gland. Urology. 2012;79(6):e77–78.
Zahir MN, Ansari TZ, Moatter T, Memon W, Pervez S. Ewing’s sarcoma arising from the adrenal gland in a young male: a case report. BMC Res Notes. 2013;6:533. doi:10.1186/1756-0500-1186-1533.
Sasaki T, Onishi T, Yabana T, Hoshina A. Ewing’s sarcoma/primitive neuroectodermal tumor arising from the adrenal gland: a case report and literature review. Tumori. 2013;99(3):e104–106. doi:10.1700/1334.14815.
Abi-Raad R, Manetti GJ, Colberg JW, Hornick JL, Shah JG, Prasad ML. Ewing sarcoma/primitive neuroectodermal tumor arising in the adrenal gland. Pathol Int. 2013;63(5):283–6.
Blas JV, Smith ML, Wasif N, Cook CB. Schlinkert RT. Ewing sarcoma of the adrenal gland: a rare entity. BMJ Case Rep. 2013;2013:bcr2012007753. doi:10.1136/bcr-2012-007753.
Dutta D, Shivaprasad KS, Das RN, Ghosh S, Chowdhury S. Primitive neuroectodermal tumor of adrenal: Clinical presentation and outcomes. J Cancer Res Ther. 2013;9(4):709–11.
Phukan C, Nirmal TJ, Kumar RM, Kekre NS. Peripheral primitive neuroectodermal tumor of the adrenal gland: A rare entity. Indian J Urol. 2013;29(4):357–9.
Yamamoto T, Takasu K, Emoto Y, Umehara T, Ikematsu K, Shikata N, Iino M, Matoba R. Latent adrenal Ewing sarcoma family of tumors: A case report. Leg Med. 2013;15(2):96–8.
Tsang YP, Lang BH, Tam SC, Wong KP. Primitive neuroectodermal adrenal gland tumour. Hong Kong Med J. 2014;20(5):444–6.
Yoon JH, Kim H, Lee JW, Kang HJ, Park HJ, Park KD, Park B-K, Shin HY, Park JD, Park S-H, et al. Ewing Sarcoma/Peripheral Primitive Neuroectodermal Tumor in the Adrenal Gland of an Adolescent: A Case Report and Review of the Literature. J Pediatr Hematol Oncol. 2014;36(7):E456–9.
Bhatt Krutika R, Trivedi Priti P, Shah Manoj J. Adult Neuroblastoma of Adrenal gland: Two case report. The Southeast Asian Journal of Case Report and Review. 2015;4(3):1742–8.
Zhang L, Yao M, Hisaoka M, Sasano H, Gao H. Primary Ewing sarcoma/primitive neuroectodermal tumor in the adrenal gland. APMIS. 2016;124(7):624-9.
Llombart-Bosch A, Machado I, Navarro S, Bertoni F, Bacchini P, Alberghini M, Karzeladze A, Savelov N, Petrov S, Alvarado-Cabrero I, et al. Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009;455(5):397–411.
Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzerkuntschik M. MIC2 is a specific marker for ewings-sarcoma and peripheral primitive neuroectodermal tumors - evidence for a common histogenesis of ewings-sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67(7):1886–93.
Gerald WL, Ladanyi M, de Alava E, Cuatrecasas M, Kushner BH, LaQuaglia MP, Rosai J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its variants. J Clin Oncol. 1998;16(9):3028–36.
Devaney K, Vinh TN, Sweet DE. Small-cell osteosarcoma of bone - an immunohistochemical study with differential diagnostic considerations. Hum Pathol. 1993;24(11):1211–25.
Riopel M, Dickman PS, Link MP, Perlman EJ. MIC2 Analysis In Pediatric Lymphomas And Leukemias. Hum Pathol. 1994;25(4):396–9.
Mhawech-Fauceglia P, Herrmann FR, Bshara W, Odunsi K, Terracciano L, Sauter G, Cheney RT, Groth J, Penetrante R. Friend leukaemia integration-1 expression in malignant and benign tumours: a multiple tumour tissue microarray analysis using polyclonal antibody. J Clin Pathol. 2007;60(6):694–700.
Saxena R, Sait S, Mhawech-Fauceglia P. Ewing sarcoma/primitive neuroectodermal tumor of the kidney: a case report. Diagnosed by immunohistochemistry and molecular analysis. Ann Diagn Pathol. 2006;10(6):363–6.
Zhong J, Chen N, Chen X, Gong J, Nie L, Xu M, Zhou Q. Peripheral primitive neuroectodermal tumor of the kidney in a 51-year-old female following breast cancer: A case report and review of the literature. Oncol Lett. 2015;9(1):108–12.
Pinto A, Dickman P, Parham D. Pathobiologic markers of the ewing sarcoma family of tumors: state of the art and prediction of behaviour. Sarcoma. 2011;2011:856190.
Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293(5539):2449–52.
Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288(3):C494–506.
Vural C, Uluoglu O, Akyurek N, Oguz A, Karadeniz C. The evaluation of CD99 immunoreactivity and EWS/FLI1 translocation by fluorescence in situ hybridization in central PNETs and Ewing’s sarcoma family of tumors. Pathol Oncol Res. 2011;17(3):619–25.
DuBois SG, Krailo MD, Gebhardt MC, Donaldson SS, Marcus KJ, Dormans J, Shamberger RC, Sailer S, Nicholas RW, Healey JH, et al. Comparative Evaluation of Local Control Strategies in Localized Ewing Sarcoma of Bone A Report From the Children’s Oncology Group. Cancer. 2015;121(3):467–75.
Acknowledgments
The authors thank the patient and his families for allowing us to publish this case report. We also thank Professor Meishan Jin (Department of Pathology, First Hospital of Jilin University, Changchun 130021, China) as the pathologist for reviewing and confirming the histological diagnosis for our patient.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author on reasonable request.
Authors’ contributions
HG and SQC wrote the manuscript and made the revisions. SKL, KXW, EPL and FPL participated in data collection. YCH collected cases and do the check. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. The data do not contain any information that could identify the patient. A copy of the written consent is available for review by the editor of this journal.
Ethics approval and consent to participate
All procedures were approved by the Ethics Committee of First Hospital of Jilin University
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, H., Chen, S., Liu, S. et al. Rare adrenal gland incidentaloma: an unusual Ewing’s sarcoma family of tumor presentation and literature review. BMC Urol 17, 24 (2017). https://doi.org/10.1186/s12894-017-0217-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-017-0217-3