
Abstract
Background
We systematically reviewed the effectiveness of intensive treatment strategies in achieving remission in patients with both early and established Rheumatoid Arthritis (RA).
Methods
A systematic literature review and meta-analysis evaluated trials and comparative studies reporting remission in RA patients treated intensively with disease modifying anti-rheumatic drugs (DMARDs), biologics and Janus Kinase (JAK) inhibitors. Analysis used RevMan 5.3 to report relative risks (RR) in random effects models with 95% confidence intervals (CI).
Results
We identified 928 publications: 53 studies were included (48 superiority studies; 6 head-to-head trials). In the superiority studies 3013/11259 patients achieved remission with intensive treatment compared with 1211/8493 of controls. Analysis of the 53 comparisons showed a significant benefit for intensive treatment (RR 2.23; 95% CI 1.90, 2.61). Intensive treatment increased remissions in both early RA (23 comparisons; RR 1.56; 1.38, 1.76) and established RA (29 comparisons RR 4.21, 2.92, 6.07). All intensive strategies (combination DMARDs, biologics, JAK inhibitors) increased remissions. In the 6 head-to-head trials 317/787 patients achieved remission with biologics compared with 229/671 of patients receiving combination DMARD therapies and there was no difference between treatment strategies (RR 1.06; 0.93. 1.21). There were differences in the frequency of remissions between early and established RA. In early RA the frequency of remissions with active treatment was 49% compared with 34% in controls. In established RA the frequency of remissions with active treatment was 19% compared with 6% in controls.
Conclusions
Intensive treatment with combination DMARDs, biologics or JAK inhibitors increases the frequency of remission compared to control non-intensive strategies. The benefits are seen in both early and established RA.
Similar content being viewed by others


Background
Remission has become a key treatment goal in rheumatoid arthritis (RA). Achieving remission with drug treatment is recommended in many clinical management guidelines [1,2,3,4,5,6]. It is also a central feature of the “treat-to-target” initiative [7, 8]. Patients who achieve remission have less disability and better quality of life than those with persisting inflammatory disease [9]. In early RA remission is particularly important due to the ‘window of opportunity’ during which early intensive treatment can halt or substantially reduce subsequent disease progression [10].
There are several definitions of remission in RA. The 2010 European League Against Rheumatism (EULAR) and American College of Rheumatology (ACR) criteria provided a framework for considering these different definitions [11]. A variety of composite measures are used to determine the presence of remission. These include the Disease Activity Score (DAS) and the Disease Activity Score for 28 joints (DAS28), the Simple Disease Activity Score (SDAI) and the Clinical Disease Activity Score (CDAI) [12,13,14]. DAS28 remission criteria have been used most frequently in trials of intensive treatments in RA, though there has been debate whether it is ideal [15].
Several systematic reviews have reported on treatment remissions in RA [16,17,18,19,20], patients likely to achieve remission [21, 22] and the strength of the rationale for treatment to target approaches in RA [23, 24]. The balance of evidence from these reviews is that intensive treatment increases remission. However, several uncertainties need to be resolved. Firstly, the relative merits of intensive treatment in early RA compared to established disease need to be considered. Secondly, it is important to know whether treatment with one type of therapy, such as biologics like tumour necrosis factor (TNF) inhibitors, will lead to more remissions than treatment with combinations of conventional disease modifying anti-rheumatic drugs (DMARDs) Finally it is important to know if one or other treatment strategy is preferable in early or established disease.
We have systematically reviewed RA clinical trials that report remissions. We evaluated both trials that compare an intensive treatment strategy with standard care and also head-to-head trials of different intensive treatment strategies. We analysed trials in early and established RA separately, taking the division between these groups as usually being 12 months since diagnosis.
Methods
Inclusion and exclusion criteria
The inclusion criteria were: randomized controlled trials or open label non-randomised comparative studies with at least one intensive treatment arm and one control arm; adult patients with RA; studies of at least 6 months duration; studies enrolling at least 50 patients; studies reporting remissions; studies using treatments in their licensed indication for RA. The intensive treatment arms used drugs considered more intensive than DMARD monotherapy. These included combination DMARDs (which could involve using short-term regular doses of steroids to control synovitis), TNF inhibitors, non-TNF biologics (tocilizumab, abatacept and rituximab), and Janus Kinase (JAK) inhibitors. We also noted whether studies used a treat-to-target approach with intensive treatments. Studies either compared one intensive treatment strategy against standard care or two different intensive treatment strategies (such as combination DMARDs and TNF inhibitors with DMARDs). Foreign language papers and published conference abstracts were excluded. Trials comparing similar types of treatment, such as two intensive DMARD regimens, were also excluded. The search identified publications from 1st January 2000 to 30th April 2017.
Search strategy
A systematic literature search was carried out using EMBASE, OVID Medline as well as hand searching the systematic reviews relevant to this topic found in the Cochrane library database. The key word search terms used were ‘arthritis, rheumatoid’ (MeSH), ‘clinical trial’ [Publication Type] (MeSH), randomised controlled trial [Publication Type] (MeSH), open label (free text) and ‘remission’ (free text). These were searched separately and in combination. The EMBASE search terms included ‘arthritis, rheumatoid’ (MeSH) all subheadings and FOCUS function, clinical trial (MeSH) Explode function.
Data collection
Two researchers (CH, DLS) independently assessed studies for eligibility and extracted data. This included year of publication, disease duration, number of treatment groups, study design, control and intensive treatment regimens, study size, remissions and study end-points. The numbers of patients achieving disease remission at the trial end-point was defined by Disease Activity Scores (DAS) < 1.6, DAS28 < 2.6 or equivalent criteria. The trials were classified as early (generally with disease durations < 1 year) or established (generally with disease durations > 1 year) reflecting the trial investigators assessments. When there were differences between assessors, they reviewed the reports together and came to a joint conclusion.
Assessing Bias
A quality assessment was completed for each paper using the Cochrane Collaboration tool for assessing risk of bias [25]. The types of bias assessed were: random sequence generation, selection bias, performance bias, detection bias, attrition bias, reporting bias and other bias (such as pharmaceutical funding). The risk was defined as low or high. We also used funnel plots to assess publication bias and associated issues [26].
Statistical analysis
Results were analysed using Review Manager 5.3 (Cochrane Collaboration, Oxford, UK). The random effects model based on DerSimonian and Laird’s method [27] was used to estimate the pooled effect sizes; this gives more equal weighting to studies of different precision in comparison with a simple inverse variance weighted approach, so accommodating between study heterogeneity. For all meta-analyses, we performed Cochrane’s chi-squared test to assess between study heterogeneity and quantified I2 statistics [25]. P-values < 0.05 were considered significant.
Some of the randomised controlled trials had more than two treatment arms: when there were two control groups the results were combined; when there were two or more intensive treatment groups only those reporting licensed dosage regimens were included.
Results
Study selection
We identified 928 publications: 440 were duplicated studies and 414 were excluded after reviewing abstracts. Seventy four full text papers were reviewed in detail; 21 were excluded and 53 selected for inclusion (Fig. 1). These papers comprised 48 superiority trials, in which an intensive treatment strategy was compared with a less intensive strategy, and 6 head-to-head trials comparing combination DMARDs with biologic treatments. The BeST paper is included in both of these groups.

PRISMA Diagram Outlining Search Strategy
Characteristics of included studies
Twenty two superiority trials evaluated patients reported as having early RA. Their maximum disease durations ranged from 3 months to 3 years. Mean or median disease durations, reported in 20 of these trials, ranged from 1 to 11 months (mean 6 months). Four of these trials studied patients with very early disease, less than 6 months from diagnosis. One trial had two different intensive treatment arms (combination DMARDs and biologics) which were both included. Six trials had two or three intensive treatment arms: in three trials biologic monotherapy treatment arms were omitted; in another three trials only licensed combination regimens were included.
Twenty six superiority trials evaluated patients with established RA. Six of these trials specified maximum disease durations (from 5 to 20 years). Mean or median disease durations, reported in all of these trials, ranged from 1 to 12 years (mean 8 years). One trial had two control groups (methotrexate or sulfasalazine monotherapy) and these were combined. Sixteen trials had two or more intensive treatment arms: three had two different licensed intensive treatments (biologics and JAK inhibitors) which were both included; in one trial the biologic monotherapy treatment arm was omitted; in a further 12 trials only licensed combination regimens were included.
Overall 19,752 RA patients were studied: 7300 in early RA and 12,452 with established RA (Table 1). There were 46 conventional RCTs, one was open label and one quasi-experimental. Twenty four trials had 2-arms, 17 had 3-arms and 7 had over three arms. The trials often reported outcomes at several different time-points, but their primary outcomes were reported at 6 months in 21 trials, at 12 months in 19 trials and at longer intervals in 8 trials (2 at 18 months, 5 at 24 months and 1 at 36 months).
DAS28 remissions (DAS28 < 2.6) were reported in 38/48 superiority trials and 4/6 head-to-head trials. DAS remissions (DAS < 1.6) were reported in 5/48 superiority trials and 2/6 head-to-head trials. Five superiority trials reported other remissions (using SDAI in 3 and unique study-specific criteria in 2). In addition, 12 superiority trials reported some or all of the new EULAR/ACR remission criteria.
Treat-to-target strategies were included within 8/48 superiority trials and 3/6 head-to-head trials, though there were substantial differences in how these strategies were delivered.
Remission in superiority trials
Overall in the 48 trials 3013/11,259 patients achieved remission with intensive treatment compared with 1211/8493 patients receiving non-intensive therapy (Table 2). Analysis of the 53 comparisons in these trials using the random effects relative risk model showed there was a highly significant benefit for intensive treatment (RR 2.23; 95% CI 1.90, 2.61). There was marked heterogeneity between studies; I2 was 84%.
In the 38 trials (40 comparisons) reporting DAS28 remissions the random risk ratio was 2.26 (95% CI 1.89, 2.71); in the 10 trials (12 comparisons) reporting other remission criteria the random risk ratio was 2.13 (95% CI 1.53, 2.98). The random risk ratios showed significant effects with trials of 6 months, 12 months and longer durations. Although the random ratio was somewhat higher in trials of 6 months duration, 17/21 trials (20/24 comparisons) were in established RA and in these the random risk ratio was 4.82 (95% CI 2.85, 8.13); in the 4 trials (4 comparisons) lasting 6 months in early RA the random risk ratio was 1.94 (95% CI 1.21, 3.11). In the 8 trials (9 comparisons) involving TTT strategies as part of intensive treatment the random risk ratio was 1.62 (95% CI 1.30, 2.03).
In the 22 trials in early RA with intensive treatments trials with 1756/3993 patients achieved remission with intensive treatment compared with 903/3307 patients receiving monotherapy. One trial evaluated two intensive treatment regimens and there were consequently 23 comparisons; 13 evaluated TNF inhibitors, 5 evaluated other biologics and 5 evaluated combination DMARDs. Analysis of the 23 comparisons in these trials showed a significant overall benefit for intensive treatment (RR 1.56; 95% CI 1.38, 1.76). There was marked heterogeneity in these studies; I2 was 74% (Table 2). A funnel plot showed a symmetrical pattern in these trials (result not shown). Four trials enrolled patients with disease durations no more than 6 months and these showed a similar benefit for intensive treatment (RR 1.47; 95%CI 1.03, 2.10) Comparison of the different intensive treatment regimens in early RA patients showed similar impacts of different intensive treatments; these ranged from a random risk ratio of 1.43 with TNF inhibitors to 2.00 with other biologics. TTT strategies also increased remissions with a random risk ratio of 1.51.
In the 26 established RA trials 1257/7266 patients achieved remission with intensive treatment compared with 308/5186 patients receiving monotherapy. Three trials evaluated two intensive treatment regimens and consequently there were 29 comparisons: 10 evaluated TNF inhibitors, 10 evaluated other biologics, 3 evaluated combination DMARDs and 6 evaluated JAK inhibitors. Analysis of these 29 comparisons trials showed a significant overall benefit for intensive treatment (RR 4.21; 95% CI 2.92, 6.07). There was marked heterogeneity in these studies; I2 was 86% (Table 2). A funnel plot showed an asymmetrical pattern in these trials (result not shown). Comparison of the different intensive treatment regimens in established RA patients showed some differences in the magnitude of effects; random risk ratios ranged from 2.41 with combination DMARDs to 6.81 with other biologics (tocilizumab, adalimumab and rituximab); however, as the confidence intervals overlapped there was no evidence these differences were significant. Only two trials used TTT strategies and although these increase remissions the 95% confidence intervals showed the finding may not have been significant (random risk ratio 2.39; 95% CI 0.90, 6.32).
Using a fixed effects model gave similar findings. In all trials the risk ratio was 2.06 (95%CI 1.94, 2.18), in early RA trials it was 1.64 (95% CI 1.54, 1.74) and in established RA the risk ratio was 3.32 (95% CI 2.94, 3.74). Interestingly the fixed model indicated TTT strategies in established RA in two trials may have been significant (risk ratio 2.19, 95% CI 1.50, 3.19.
Remission in head to head trials
Overall in the 6 trials 317/787 patients achieved remission with TNF inhibitors compared with 229/671 of patients receiving combination DMARD therapies. Analysis of these 6 trials using the random effects relative risk model (Table 3) showed there was a no different between treatment strategies (RR 1.06; 95% CI 0.93. 1.21). There was little heterogeneity between studies; I2 was 21%. Comparing 4 early RA and 2 established RA trials separately also showed no evidence of a significant difference between groups (Table 3). However, comparisons of the first 6 months results in the two established RA trials showed more remissions with TNF inhibitors using the random effects relative risk model (RR 1.74, 95% CI 1.14, 2.64). The fixed effects model gave similar findings (RR 1.90; 95% CI 1.17, 3.10).
Frequency of remissions
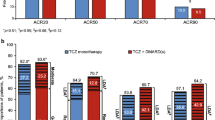
There were marked differences in the frequency of remissions in active and control groups in both early and established RA (Fig. 2). In early RA the average frequency of remissions with active treatment was 49%: in 10 early RA trials 50% or more active patients achieved remissions; the highest rate was 86% in the U-Act-Early (tocilizumab) trial and the lowest rate was 18% in the St Clair (Infliximab) trial. In early RA controls the average frequency of remission was 34%: in four trials 50% or more controls achieved remissions; and the lowest rate in controls was 18% in the Image (rituximab) trial. The average difference in remission rates between active and control group in early RA trials was 15%.

Remissions in control and active groups shown as percent patients in each group in early and established RA
In established RA the average frequency of remissions with active treatment was 19%: in only one trial did 50% or more active patients achieved remission (65% in the Ticora trial of combination DMARDs); in 14 trials 10% or less active patients achieved remission and, in the Reflex, (rituximab) and RA Beam (baricitinib and adalimumab) trials only 3% of patients achieved remissions. In established RA controls the average frequency of remission was 6%: in 22 trials less than 5% of controls achieved remissions; and in the Reflex (rituximab) trial no control patient achieved an end-point remission. The average difference in remission rates between active and control group in early RA trials was 13%.
Quality and risk of Bias
Quality assessment, using the Cochrane Collaboration tool for assessing risk of bias, showed overall quality was high with low risks of bias (Table 1).
Discussion
TNF Inhibitors, other biologics and combination DMARDS were all effective in increasing remission in early and established RA. Treat to target strategies, which usually involved intensive DMARDs, were also effective. JAK inhibitors were similarly effective in established RA; there was no data about their impact in early disease. Although other biologics achieved numerically higher risk ratios in both early and established RA the overlapping confidence intervals gave no support to the view that these differences are clinically significant. The benefits of different types of intensive treatment were therefore broadly similar. Trials of varying durations, from 6 months to more than 12 months, all showed intensive treatments increased remissions. There was no evidence that patients with very early RA of no more than 6 months disease duration benefited more from intensive treatments. We excluded trials with durations of less than 6 months to ensure we did not disadvantage the assessment of intensive treatment strategies using slower acting DMARDs. The head-to-head trials supported the similarities between treatments with combination DMARD strategies and TNF inhibitor strategies, which achieved similar end-point remission rates. There was however, some evidence that TNF inhibitors increased the early remission rates, in keeping with their relatively rapid onset of action compared to conventional DMARD combinations.
TNF Inhibitors, other biologics and combination DMARDS were all effective in increasing remission in early and established RA. There was no evidence that patients with very early RA of no more than 6 months disease duration benefited more from intensive treatments. JAK inhibitors were similarly effective in established RA; there was no data about their impact in early disease. Although other biologics achieved numerically higher risk ratios in both early and established RA the overlapping confidence intervals gave no support to the view that these differences are clinically significant. The head-to-head trials supported the similarities between treatments with combination DMARD strategies and TNF inhibitor strategies, which achieved similar end-point remission rates. There was however, some evidence that TNF inhibitors increased the early remission rates, in keeping with their relatively rapid onset of action compared to conventional DMARD combinations.
The overall quality of the studies was relatively high. However, there was evidence of marked heterogeneity in their findings with most comparisons having high I2 values. This heterogeneity meant that in some intensive treatment arms in early RA over 70% patients achieved remission while in other intensive treatment arms in established RA under 10% patients achieved remission. These differences are likely to reflect patient selection more than treatment efficacy, with very early RA patients having no previous DMARDs are highly likely to achieve remission with intensive treatment while established RA patients who have failed multiple prior treatments are unlikely to do so.
The most likely explanation for the asymmetrical funnel plot in trials in established RA relates to specifically including studies using treatments in their licensed indication which were published between 2000 and 2017. A consequence is that potential intensive treatments which were evaluated in RA patients but were not found to be effective, were not included. Firstly, small initial studies with new drugs which would have shown negative results for remissions were not included as the treatments were never licensed for RA. An example is the spleen tyrosine kinase inhibitors [81]. Secondly, some TNF inhibitors were not effective in RA and were therefore not licensed; an example is Lenercept, which failed to show sustained benefit in clinical trials [82]. Finally, combinations with DMARDs were tried in the 1980’s, before remission was measured or reported; these trials were mainly negative [83]; subsequent trials of intensive DMARD combinations reporting remission which were published after 2000 studied treatments which were known to be effective in combination. These factors mean the funnel plot of remissions in established RA would not include small trials with negative findings because of the selection criteria used. As this report focuses on the benefits of different intensive treatment strategies using licensed treatments given at their approved dosages we do not think an asymmetric funnel plot changes our conclusions.
Our systematic review has a number of limitations. Firstly, studies not reporting remission data were excluded, though they often show clinically important improvements with intensive treatment. Secondly, studies reported remissions differently; for example, DAS and DAS28 remissions are similar but not identical. Thirdly, studies were of variable duration and comparing remission rates over 6 and 12 months or more is not ideal; however, variations in treatments and patient selection meant there was no evidence for one particular time point being best. Fourthly, studies differed in the way they handled non-responders, with some trials stopping treatment if patients had not responded within 3 months or so and applying non-responder imputations. This approach may alter the remission rates in the non-intensive treatment by making it appear smaller than it might have been if treatment was continued. Fifthly, as mentioned previously, studies enrolled different patient groups in whom the likelihood of achieving remissions was very different. Sixthly, the intensive combination DMARD regimens used in the trials have been combined together, even though they represent a wide range of different strategies, not all of which appeared highly effective. In one study by Schipper et al. [58] some patients in active and control groups had monotherapy and others had biologics, so the trial is not just a comparison of one treatment strategy; however, excluding it made no difference to the conclusions. Finally, there is debate about the benefits of combining the results of different trials in a meta-analysis, considering their potential degrees of clinical heterogeneity. As we have also undertaken extensive sub-group analyses we consider the approach we have taken is justified in this particular clinical context.
Our results have several implications for clinical practice. Firstly, they show that intensive treatment strategies lead to more remissions than conventional care in both early and established RA. This finding is generally supportive of the treat-to-target approach currently recommended [7, 8], although we have not attempted to dissociate the impact of giving intensive treatment from the impact of the target. Secondly, they show that initial treatment with conventional DMARD combinations has similar effectiveness to early biologics. This finding is supportive of the current recommendations about biologic treatments from the National Institute for Health and Care Excellence (NICE), which recommend trying combination DMARDs before biologic treatment [84]. Thirdly, they question whether remission is necessarily the ideal target for treatment in established RA, as it is only achieved by a minority of patients in most trials of intensive treatment. There may be greater value of aiming for low disease activity states, in which case these need to be measured in future trials. The EULAR good response criteria can be used to assess the frequency of achieving low disease activity states measures using DAS28. Current guidance on treat to target includes aiming for low disease activity in some patients.
One issue this review cannot address is treatment sequencing. Some experts believe most early RA patients should receive methotrexate monotherapy initially for a few months and only have intensive treatments if they fail to respond. Other experts recommend early intensive treatment followed by treatment tapering. It is possible to find individual trials within our systematic review, which support both options, but there is no systematic evidence to support or refute either approach. One final pair of inter-related uncertainties is the optimal time to assess remission and the most suitable assessment to evaluate its presence. Combining superiority and head-to-head trials (Tables 1 and 4) shows 23 (43%) lasted 12 months, 20 (38%) lasted 6 months and 10 (19%) lasted over 12 months, with the longest (BROSG trial evaluating combination DMARDs) lasting 3 years. This finding suggests trials of 12 months or longer seem preferable. Although most trials reported DAS28 remissions, this represents an historical target and there is now greater emphasis on stricter remission criteria.
Conclusions
Intensive treatment with combination DMARDs, biologics or JAK inhibitors increases the frequency of remission compared to control non-intensive strategies. The benefits are seen in both early and established RA. The relative merits of different remission criteria in trials is a complex question but changing criteria has the disadvantage of making it difficult to compare trials with newer criteria and those using more historic methods.
Abbreviations
- ACR:
-
American College of Rheumatology
- BROSG:
-
British rheumatoid outcome study group
- CDAI:
-
Clinical disease activity score
- CI:
-
Confidence intervals
- DAS:
-
Disease activity score
- DAS28:
-
Disease activity score for 28 joints
- DMARDs:
-
Disease modifying anti rheumatic drugs
- EULAR:
-
European league against rheumatism
- JAK inhibitors:
-
Janus kinase inhibitors
- NICE:
-
National Institute for Health and Care Excellence
- RA:
-
Rheumatoid arthritis
- RR:
-
Relative risks
- SDAI:
-
Simple diseasee activity score
- TNF inhibitors:
-
Tumour necrosis factor inhibitors
- TTT:
-
Treat to target
References
Smolen JS, Landewe R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77.
Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26.
Gaujoux-Viala C, Gossec L, Cantagrel A, Dougados M, Fautrel B, Mariette X, et al. Recommendations of the French Society for Rheumatology for managing rheumatoid arthritis. Joint Bone Spine. 2014;81:287–97.
Albrecht K, Kruger K, Wollenhaupt J, Alten R, Backhaus M, Baerwald C, et al. German guidelines for the sequential medical treatment of rheumatoid arthritis with traditional and biologic disease-modifying antirheumatic drugs. Rheumatol Int. 2014;34:1–9.
Bykerk VP, Akhavan P, Hazlewood GS, Schieir O, Dooley A, Haraoui B, et al. Canadian rheumatology association recommendations for pharmacological management of rheumatoid arthritis with traditional and biologic disease-modifying antirheumatic drugs. J Rheumatol. 2012;39:1559–82.
(UK) NCCfCC. Rheumatoid arthritis: national clinical guideline for management and treatment in adults: National institute for health and clinical excellence: guidance. National collaborating centre for chronic conditions (UK). London: Royal College of Physicians (UK); 2009.
Smolen JS, Breedveld FC, Burmester GR, Bykerk V, Dougados M, Emery P, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis. 2016;75:3–15.
Huizinga T, Knevel R. Rheumatoid arthritis: 2014 treat-to-target RA recommendations--strategy is key. Nat Rev Rheumatol. 2015;11:509–11.
Alemao E, Joo S, Kawabata H, Al MJ, Allison PD, Rutten-van Molken MP, et al. Effects of achieving target measures in rheumatoid arthritis on functional status, quality of life, and resource utilization: analysis of clinical practice data. Arthritis Care Res. 2016;68:308–17.
van Nies JA, Tsonaka R, Gaujoux-Viala C, Fautrel B, van der Helm-van Mil AH. Evaluating relationships between symptom duration and persistence of rheumatoid arthritis: does a window of opportunity exist? Results on the Leiden early arthritis clinic and ESPOIR cohorts. Ann Rheum Dis. 2015;74:806–12.
Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European league against rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Ann Rheum Dis. 2011;70:404–13.
Prevoo ML, van Gestel AM, van THMA, van Rijswijk MH, van de Putte LB, van Riel PL. Remission in a prospective study of patients with rheumatoid arthritis. American rheumatism association preliminary remission criteria in relation to the disease activity score. Br J Rheumatol. 1996;35:1101–5.
Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology. 2003;42:244–57.
Aletaha D, Ward MM, Machold KP, Nell VP, Stamm T, Smolen JS. Remission and active disease in rheumatoid arthritis: defining criteria for disease activity states. Arthritis Rheum. 2005;52:2625–36.
Sheehy C, Evans V, Hasthorpe H, Mukhtyar C. Revising DAS28 scores for remission in rheumatoid arthritis. Clin Rheumatol. 2014;33:269–72.
Schipper LG, van Hulst LT, Grol R, van Riel PL, Hulscher ME, Fransen J. Meta-analysis of tight control strategies in rheumatoid arthritis: protocolized treatment has additional value with respect to the clinical outcome. Rheumatology. 2010;49:2154–64.
Knevel R, Schoels M, Huizinga TW, Aletaha D, Burmester GR, Combe B, et al. Current evidence for a strategic approach to the management of rheumatoid arthritis with disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2010;69:987–94.
Jurgens MS, Welsing PM, Jacobs JW. Overview and analysis of treat-to-target trials in rheumatoid arthritis reporting on remission. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S56–63.
Bykerk VP, Keystone EC, Kuriya B, Larche M, Thorne JC, Haraoui B. Achieving remission in clinical practice: lessons from clinical trial data. Clin Exp Rheumatol. 2013;31:621–32.
Stoffer MA, Schoels MM, Smolen JS, Aletaha D, Breedveld FC, Burmester G, et al. Evidence for treating rheumatoid arthritis to target: results of a systematic literature search update. Ann Rheum Dis. 2016;75:16–22.
Hamann P, Holland R, Hyrich K, Pauling JD, Shaddick G, Nightingale A, et al. Factors associated with sustained remission in rheumatoid arthritis in patients treated with anti-tumor necrosis factor. Arthritis Care Res. 2017;69:783–93.
Katchamart W, Johnson S, Lin HJ, Phumethum V, Salliot C, Bombardier C. Predictors for remission in rheumatoid arthritis patients: a systematic review. Arthritis Care Res. 2010;62:1128–43.
Pincus T, Castrejon I, Bergman MJ, Yazici Y. Treat-to-target: not as simple as it appears. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S10–20.
Solomon DHBA, Katz JN, Radner H, Brown EM, Fraenkel L. Review: treat to target in rheumatoid arthritis: fact, fiction, or hypothesis? Arthritis Rheumatol. 2014;66:775–82.
Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from http://handbook.cochrane.org.
Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ. 2011;343:d4002.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88.
Atsumi T, Yamamoto K, Takeuchi T, Yamanaka H, Ishiguro N, Tanaka Y, et al. The first double-blind, randomised, parallel-group certolizumab pegol study in methotrexate-naive early rheumatoid arthritis patients with poor prognostic factors, C-OPERA, shows inhibition of radiographic progression. Ann Rheum Dis. 2016;75:75–83.
Bakker MF, Jacobs JW, Welsing PM, Verstappen SM, Tekstra J, Ton E, et al. Low-dose prednisone inclusion in a methotrexate-based, tight control strategy for early rheumatoid arthritis: a randomized trial. Ann Intern Med. 2012;156:329–39.
Bijlsma JWJ, Welsing PMJ, Woodworth TG, Middelink LM, Petho-Schramm A, Bernasconi C, et al. Early rheumatoid arthritis treated with tocilizumab, methotrexate, or their combination (U-act-early): a multicentre, randomised, double-blind, double-dummy, strategy trial. Lancet. 2016;388:343–55.
Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54:26–37.
Burmester GR, Rigby WF, van Vollenhoven RF, Kay J, Rubbert-Roth A, Kelman A, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75:1081–91.
Capell HA, Madhok R, Porter DR, Munro RA, McInnes IB, Hunter JA, et al. Combination therapy with sulfasalazine and methotrexate is more effective than either drug alone in patients with rheumatoid arthritis with a suboptimal response to sulfasalazine: results from the double-blind placebo-controlled MASCOT study. Ann Rheum Dis. 2007;66:235–41.
Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–806.
Detert J, Bastian H, Listing J, Weiss A, Wassenberg S, Liebhaber A, et al. Induction therapy with adalimumab plus methotrexate for 24 weeks followed by methotrexate monotherapy up to week 48 versus methotrexate therapy alone for DMARD-naive patients with early rheumatoid arthritis: HIT HARD, an investigator-initiated study. Ann Rheum Dis. 2013;72:844–50.
Dougados M, Kissel K, Sheeran T, Tak PP, Conaghan PG, Mola EM, et al. Adding tocilizumab or switching to tocilizumab monotherapy in methotrexate inadequate responders: 24-week symptomatic and structural results of a 2-year randomised controlled strategy trial in rheumatoid arthritis (ACT-RAY). Ann Rheum Dis. 2013;72:43–50.
Emery PBG, Bykerk VP, Combe BG, Furst DE, Barre E, Karyekar CS, et al. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month double-blind treatment period. Ann Rheum Dis. 2015;74:19–26.
Emery P, Breedveld FC, Hall S, Durez P, Chang DJ, Robertson D, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet. 2008;372:375–82.
Emery P, Fleischmann RM, Moreland LW, Hsia EC, Strusberg I, Durez P, et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009;60:2272–83.
Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–23.
Emery P, Deodhar A, Rigby WF, Isaacs JD, Combe B, Racewicz AJ, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (study evaluating Rituximab's efficacy in MTX iNadequate rEsponders (SERENE)). Ann Rheum Dis. 2010;69:1629–35.
Emery P, Bingham CO 3rd, Burmester GR, Bykerk VP, Furst DE, Mariette X, et al. Certolizumab pegol in combination with dose-optimised methotrexate in DMARD-naive patients with early, active rheumatoid arthritis with poor prognostic factors: 1-year results from C-EARLY, a randomised, double-blind, placebo-controlled phase III study. Ann Rheum Dis. 2017;76:96–104.
Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, al XL. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243–52.
Genovese MC, McKay JD, Nasonov EL, Mysler EF, da Silva NA, Alecock E, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58:2968–80.
Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, van Zeben D, Kerstens PJ, Hazes JM, et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum. 2005;52:3381–90.
Grigor C, Capell H, Stirling A, McMahon AD, Lock P, Vallance R, et al. Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet. 2004;364:263–9.
Hetland ML, Stengaard-Pedersen K, Junker P, Lottenburger T, Ellingsen T, Andersen LS, et al. Combination treatment with methotrexate, cyclosporine, and intraarticular betamethasone compared with methotrexate and intraarticular betamethasone in early active rheumatoid arthritis: an investigator-initiated, multicenter, randomized, double-blind, parallel-group, placebo-controlled study. Arthritis Rheum. 2006;54:1401–9.
Horslev-Petersen K, Hetland ML, Junker P, Podenphant J, Ellingsen T, Ahlquist P, et al. Adalimumab added to a treat-to-target strategy with methotrexate and intra-articular triamcinolone in early rheumatoid arthritis increased remission rates, function and quality of life. The OPERA study: an investigator-initiated, randomised, double-blind, parallel-group, placebo-controlled trial. Ann Rheum Dis. 2014;73:654–61.
Kavanaugh A, Fleischmann RM, Emery P, Kupper H, Redden L, Guerette B, et al. Clinical, functional and radiographic consequences of achieving stable low disease activity and remission with adalimumab plus methotrexate or methotrexate alone in early rheumatoid arthritis: 26-week results from the randomised, controlled OPTIMA study. Ann Rheum Dis. 2013;72:64–71.
Kivitz A, Olech E, Borofsky M, Zazueta BM, Navarro-Sarabia F, Radominski SC Bao M, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res. 2014;66:1653–61.
Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363:675–81.
Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52:2263–71.
Kremer JM, Blanco R, Brzosko M, Burgos-Vargas R, Halland AM, Vernon E, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63:609–21.
Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–81.
Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E, et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med. 2013;159:253–61.
Nam JL, Villeneuve E, Hensor EM, Wakefield RJ, Conaghan PG, Green MJ, et al. A randomised controlled trial of etanercept and methotrexate to induce remission in early inflammatory arthritis: the EMPIRE trial. Ann Rheum Dis. 2014;73:1027–36.
Nam JL Villeneuve EHE, Hensor EM, Conaghan PG, Keen HI, Buch MH, Gough AK, Green MJ, et al. Remission induction comparing infliximab and high-dose intravenous steroid, followed by treat-to-target: a double-blind, randomised, controlled trial in new-onset, treatment-naive, rheumatoid arthritis (the IDEA study). Ann Rheum Dis. 2014;73:75–85.
Schiff M, Keiserman M, Codding C, Songcharoen S, Berman A, Nayiager S, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-Centre, randomised, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67:1096–103.
Schipper LG, Vermeer M, Kuper HH, Hoekstra MO, Haagsma CJ, Den Broeder AA, et al. A tight control treatment strategy aiming for remission in early rheumatoid arthritis is more effective than usual care treatment in daily clinical practice: a study of two cohorts in the Dutch rheumatoid arthritis monitoring registry. Ann Rheum Dis. 2012;71:845–50.
Smolen JS, Emery P, Ferraccioli GF, Samborski W, Berenbaum F, Davies OR, et al. Certolizumab pegol in rheumatoid arthritis patients with low to moderate activity: the CERTAIN double-blind, randomised, placebo-controlled trial. Ann Rheum Dis. 2015;74:843–50.
Smolen JS, Kay J, Doyle MK, Landewe R, Matteson EL, Wollenhaupt J, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet. 2009;374:210–21.
Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97.
Smolen J, Landewe RB, Mease P, Brzezicki J, Mason D, Luijtens K, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68:797–804.
Soubrier M, Puechal X, Sibilia J, Mariette X, Meyer O, Combe B, et al. Evaluation of two strategies (initial methotrexate monotherapy vs its combination with adalimumab) in management of early active rheumatoid arthritis: data from the GUEPARD trial. Rheumatology. 2009;48:1429–34.
St Clair EW, van der Heijde DM, Smolen JS, Maini RN, Bathon JM, Emery P, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50:3432–43.
Symmons D, Tricker K, Harrison M, Roberts C, Davis M, Dawes P, et al. Patients with stable long-standing rheumatoid arthritis continue to deteriorate despite intensified treatment with traditional disease modifying anti-rheumatic drugs--results of the British rheumatoid outcome study group randomized controlled clinical trial. Rheumatology. 2006;45:558–65.
Tak PP, Rigby WF, Rubbert-Roth A, Peterfy CG, van Vollenhoven RF, Stohl W, et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis. 2011;70:39–46.
Takeuchi T, Yamanaka H, Ishiguro N, Miyasaka N, Mukai M, Matsubara T, et al. Adalimumab, a human anti-TNF monoclonal antibody, outcome study for the prevention of joint damage in Japanese patients with early rheumatoid arthritis: the HOPEFUL 1 study. Ann Rheum Dis. 2014;73:536–43.
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:652–62.
van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–70.
van Eijk IC, Nielen MM, van der Horst-Bruinsma I, Tijhuis GJ, Boers M, Dijkmans BA, et al. Aggressive therapy in patients with early arthritis results in similar outcome compared with conventional care: the STREAM randomized trial. Rheumatology. 2012;51:686–94.
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, Garcia Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
Verstappen SM, Jacobs JW, van der Veen MJ, Heurkens AH, Schenk Y, ter Borg EJ, et al. Intensive treatment with methotrexate in early rheumatoid arthritis: aiming for remission. Computer assisted Management in Early Rheumatoid Arthritis (CAMERA, an open-label strategy trial). Ann Rheum Dis. 2007;66:1443–9.
Weinblatt ME, Bingham CO 3rd, Mendelsohn AM, Kim L, Mack M, Lu J, Baker D, et al. Intravenous golimumab is effective in patients with active rheumatoid arthritis despite methotrexate therapy with responses as early as week 2: results of the phase 3, randomised, multicentre, double-blind, placebo-controlled GO-FURTHER trial. Ann Rheum Dis. 2013;72:381–9.
Westhovens R, Robles M, Ximenes AC, Nayiager S, Wollenhaupt J, Durez P, et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann Rheum Dis. 2009;68:1870–7.
Heimans L, Wevers-de Boer KV, Visser K, Goekoop RJ, van Oosterhout M, Harbers JB, et al. A two-step treatment strategy trial in patients with early arthritis aimed at achieving remission: the IMPROVED study. Ann Rheum Dis. 2014;73:1356–61.
Leirisalo-Repo M, Kautiainen H, Laasonen L, Korpela M, Kauppi MJ, Kaipiainen-Seppanen O, et al. Infliximab for 6 months added on combination therapy in early rheumatoid arthritis: 2-year results from an investigator-initiated, randomised, double-blind, placebo-controlled study (the NEO-RACo study). Ann Rheum Dis. 2013;72:851–7.
O'Dell JR, Mikuls TR, Taylor TH, Ahluwalia V, Brophy M, Warren SR, et al. Therapies for active rheumatoid arthritis after methotrexate failure. N Engl J Med. 2013;369:307–18.
Scott DL, Ibrahim F, Farewell V, O'Keeffe AG, Walker D, Kelly C, et al. Tumour necrosis factor inhibitors versus combination intensive therapy with conventional disease modifying anti-rheumatic drugs in established rheumatoid arthritis: TACIT non-inferiority randomised controlled trial. BMJ. 2015;350:h1046.
Moreland LW, O'Dell JR, Paulus HE, Curtis JR, Bathon JM, St Clair EW, et al. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the treatment of early aggressive rheumatoid arthritis trial. Arthritis Rheum. 2012;64:2824–35.
Scott DL. Role of spleen tyrosine kinase inhibitors in the management of rheumatoid arthritis. Drugs. 2011;71:1121–32.
Rau R, Sander O, van Riel P, van de Putte L, Hasler F, Zaug M, et al. Intravenous human recombinant tumor necrosis factor receptor p55-fc IgG1 fusion protein Ro 45-2081 (lenercept): a double blind, placebo controlled dose finding study in rheumatoid arthritis. J Rheumatol. 2003;30:680–90.
Felson DT, Anderson JJ, Meenan RF. The efficacy and toxicity of combination therapy in rheumatoid arthritis. A meta-analysis. Arthritis Rheum. 1994;37:1487-91.
(NICE). NIfHaCE: Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for rheumatoid arthritis not previously treated with DMARDs or after conventional DMARDs only have failed. https://www.nice.org.uk/guidance/ta375
Acknowledgements
On behalf of TITRATE Programme Investigators.
Work Stream A: Heidi Lempp, Jackie Sturt and Louise Prothero;
Work Stream B: Isabel Neatrour, Rhiannon Baggott, Fowzia Ibrahim, Brian Tom, Allan Wailoo, James Galloway, Gabrielle Kingsley and David Scott.
Work Stream C: Brian Tom, Fowzia Ibrahim & David L Scott.
Funding
CH is a South Thames Rheumatology Specialist Registrar working in Kings College Hospital. FI is supported by the Academic department of Rheumatology. DLS is supported by TITRATE study programme.
The TITRATE study is funded by the National Institute for Health Research (NIHR) as one of its Programme Grants For Applied Research (Grant Reference Number: RP-PG-0610-10066; Programme Title: Treatment Intensities and Targets in Rheumatoid Arthritis Therapy: Integrating Patients’ And Clinicians’ Views – The TITRATE Programme. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social care.
Availability of data and materials
Data generated during the review will be available from the first author on request.
Author information
Authors and Affiliations
Consortia
Contributions
CH reviewed the articles, extracted data and drafted the manuscript. FI & DS participated in data extraction and commented on draft manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval was not required for this systematic review, as it is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hughes, C.D., Scott, D.L., Ibrahim, F. et al. Intensive therapy and remissions in rheumatoid arthritis: a systematic review. BMC Musculoskelet Disord 19, 389 (2018). https://doi.org/10.1186/s12891-018-2302-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-018-2302-5