
Abstract
Purpose
Chronic intermittent hypoxia (CIH) causes lung injury but the mechanism is unclear. Ferroptosis is a novel form of programmed cell death. In this research, we attempted to explore the role of ferroptosis in CIH-induced lung injury both in vitro and in vivo.
Methods
Sprague-Dawley rats were randomly separated into control group, CIH group and CIH + ferrostatin-1 group (CIH + Fer-1). Rats in the CIH group and CIH + Fer-1 group were exposed to intermittent hypoxia for 12 weeks. Human bronchial epithelial cell line (BEAS-2B) was cultivated for 24 h in either conventional culture medium or under CIH conditions. Fer-1 was applied to observe its treatment effects. Histological changes were evaluated by Hematoxylin–eosin (HE) staining and masson staining. The expression levels of Acyl-CoA synthetase long-chain family member 4 (ACSL4), glutathione peroxidase 4 (GPX4), interleukin-6 (IL-6) and tumour necrosis factor α (TNFα) were detected via qRT-PCR or Western blot. Cell counting kit-8 (CCK-8) was used to assess cell viability. The apoptotic rate and reactive oxygen species (ROS) was calculated by flow cytometry.
Results
Histology showed that CIH treatment induced lung injury and pulmonary fibrosis in lung tissue. After Fer-1 treatment, the pathological changes caused by CIH alleviated. The mRNA and protein levels of GPX4 decreased significantly in lung tissues of CIH-treated rats and BEAS-2B, (p < 0.05). The mRNA and protein levels of ACSL4 increased significantly in lung tissues of CIH-treated rats and BEAS-2B, (p < 0.05). The mRNA levels of IL-6 and TNFα in BEAS-2B increased after CIH treatment, (p < 0.05). Cell viability decreased, apoptosis rate and ROS increased in CIH-treated BEAS-2B, (p < 0.05). Cotreatment with Fer-1 reversed CIH-induced apoptosis, cell viability, ROS accumulation, mRNA and protein levels of GPX4, ACSL4, IL-6 and TNFα both in vitro and in vivo (p < 0.05).
Conclusions
Ferroptosis occurred in CIH-induced lung injury, both in vitro and in vivo. The ferroptosis inhibitor Fer-1 alleviated cell injury and ferroptosis in CIH-treated BEAS-2B and lung tissues of rats.
Similar content being viewed by others

Introduction
Obstructive sleep apnea (OSA) is a chronic respiratory disease with high incidence. The prevalence of OSA has reached 20% in men and 10% in women both in Western countries and Eastern countries [1]. The pathophysiological characteristics of OSA include chronic intermittent hypoxia (CIH) induced by periodic upper airway collapse or obstruction [1]. Target organ injury in OSA patients has received widespread attention. Cardiovascular injury, insulin resistance, cerebral vascular diseases, kidney injury and non-alcoholic fatty liver disease are common complications of OSA [2,3,4,5,6]. At present, clinical evidences and animal experiments have confirmed that CIH induces lung injury and aggravates existing lung injury [7,8,9,10].
In a cohort of 5108 patients followed for 10 years, FEV1 and FVC declined more rapidly in patients with high OSA probability compared with low OSA probability [11]. The prevalence of chronic cough, asthma, chronic obstructive pulmonary disease (COPD), lung hypertension and other respiratory diseases in OSA patients was significantly increased [12, 13]. Naranjo et al. [14] reported that hospitalized COPD patients with unrecognized OSA had higher readmission and mortality rates than those without OSA. The above clinical studies suggested that OSA was a risk factor of lung injury. Previous animal studies found that eight weeks of CIH exposure induced inflammation and injury in rat lung tissue [7]. Twelve weeks of CIH exposure caused ROS aggregation, mitochondrial damage and apoptosis in rat lung tissue [8]. The molecular mechanism by which CIH causes and aggravates lung injury is, however, unclear.
Ferroptosis is a common type of cell death which is iron-regulated, characterized by lipid-peroxidation [15]. Ferroptosis has been extensively studied in recent years and is involved in the development of various ischemia-reperfusion diseases [16]. The pathophysiological characteristics of CIH are similar to ischemia-reperfusion injury. Furthermore, Chen et al. [17] found that CIH exposure induced ferroptosis in rat liver tissue. However, there was no literature to report whether ferroptosis regulated CIH-related lung injury.
This study explored the role of ferroptosis in CIH-related lung injury using cell and animal experiments. Meanwhile, the treatment effectiveness of ferroptosis inhibitor ferrostatin-1 (Fer-1) was observed both in vitro and in vivo.
Materials and methods
Experimental animals and subgroups
The study included male Sprague-Dawley rats aged 8 weeks. The rats were purchased from the Chinese Academy of Sciences' Animal Center (Shanghai, China). All rats were kept in an animal facility with a 12-h light/dark cycle, regular food and tap water. The rats were separated into control group (n = 6), CIH group (n = 6) and CIH + Fer-1 group (n = 6) via computer random number method. The rats in the CIH group and CIH + Fer-1 group were put into a computer-controlled intermittent low oxygen tank. Nitrogen was admitted to the intermittent low oxygen tank for 40 s to decrease the fractional oxygen concentration (FcO2) to 6%, stabilized at that level for 20 s, after which oxygen was introduced for the next 40 s to increase FcO2 to 21% and maintained in that state for 20 s. This intermittent low oxygen cycle was maintained for 8 h each day for 12 weeks in CIH group and CIH + Fer-1 group [7, 18, 19]. After 8 weeks of CIH treatment, the CIH + Fer-1 group were given 2 mg/kg Fer-1 via abdominal injection. After modeling, the rats were intraperitoneally injected with 50 mg/kg pentobarbital sodium, and lung tissues were harvested for the histopathological examination, masson staining, Western blot analysis and measurement of genetic expression. The Ethics Committee of the Fujian Medical University Laboratory Animal Center approved this study (approve number: IACUCFJMU2022-0031), and all rats were euthanized in accordance with ARRIVE guidelines and treated humanely.
Cell culture
Human bronchoalveolar epithelial cells (BEAS-2B), provided by Biyuntian Biological Technology Co., Ltd (Shanghai, China). The cell lines were commercial cell lines derived from normal human bronchial epithelial tissue. BEAS-2B were cultured in an incubator at 37 °C with 5% CO2. The culture medium was a mixture of DMEM (HyClone Laboratory INC., USA), 10% (vol/vol) fetal bovine serum (Gibco Life Technologies INC., USA) with penicillin (100 units/mL) and streptomycin (100 µg/mL). BEAS-2B were divided into control group, CIH group, and CIH + Fer-1 group. For the CIH group, once BEAS-2B cells reached around 75% confluency, CIH exposure was applied for 24 h by cycling between hypoxia (1% O2 with 5% CO2 balanced with N2 for 60 min) and normoxia (21% O2 with 5% CO2 balanced with N2 for 30 min). For the CIH + Fer-1 group, BEAS-2B was co-treated with 10 µM Fer-1 and CIH for 24 h.
Hematoxylin–eosin (HE) staining and masson staining of lung tissue of rats
The lung tissue was harvested and immersion-fixed in 4% polymethanol for 24 h, dehydrated in gradient ethanol and then handled to get paraffin blocks. The lung samples were sectioned into 4 µm thicknesses, stained with hematoxylin and eosin or masson trichrome, then observed under a light microscope (Olympus BX50, Tokyo, Japan).
Protein extraction and Western blot analysis
The harvested lung tissue and BEAS-2B were used to extract proteins using Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime, China). The protein concentration was determined by BCA protein concentration kit (Beyotime, China). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done in advance to separate the proteins. The proteins were subsequently electroblotted onto polyvinylidene difluoride membranes (Millipore, 150 Billerica, MA, USA). The membranes were blocked for 2 h in a solution of 5% nonfat dry milk in Tris Buffer Solution Tween (TBST). The primary antibodies ACSL4 (Acyl-CoA synthetase long-chain family member 4, Abcam, 1: 1000), GPX4 (glutathione peroxidase 4, Abcam, 1: 1000) and β-actin (Abcam, 1: 1000) were then incubated at 4 °C overnight. The membrane was washed with TBST every 10 min. Membranes were washed and stained for 1 h at room temperature with goat anti-rabbit IgG secondary antibody (Abcam, 1: 10,000). After that, the membranes were washed three times with TBST every 10 min. The blots were cut prior to hybridisation with antibodies during blotting. The bands were developed using an improved ECL kit (Thermo Scientific, Rockford, USA). GelDoc XR equipment was used to take the images, which were then analyzed using Image Lab Software (Bio-Rad). Additional file 1 is the original WB image in the manuscript.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
For RNA extraction, ground lung tissue and BEAS-2B were utilized. Total RNA was extracted using the Total RNA Isolation Kit V2 (Vazyme, China), and cDNA was produced using the HiScript III All-in-one RT SuperMix Perfect for qPCR (Vazyme, China). RT-qPCR was performed on an ABI 7500 thermocycler (Applied Biosystems, Foster City, CA, USA). The mRNA expression levels of GPX4 and ACSL4 were assessed using the SYBR Green PCR Master Mix (Vazyme). The suitable primers were summarized in Table 1. The relative expression levels of mRNA were reported as fold change compared to levels detected in controls via the ΔΔCt method.
Cell viability test
The viability of BEAS-2B cells was determined using a CCK-8 kit (Beyotime Bio Inc., China). A 96-well plate was seeded with 10,000 cells per well, with five replicate wells in each group. To minimize errors, phosphate-buffered saline (PBS) was added to the surrounding wells. After modeling, cells were incubated for 2 h at 37 °C in fresh media containing 1 mg/mL CCK-8 solution. The absorbance at 450 nm was measured using a microplate reader. Cell viability was calculated as (experimental group-blank control)/ (negative control-blank control) × 100%.
Apoptosis assay
The apoptosis rate was detected by flow cytometry. 1 × 105 cells were collected per well after modeling, with three replicate wells in each group. Cells were resuspended in 200 µl binding buffer with 10 µl PI and 5 µl Annexin V-FITC after being washed with PBS. A C6 flow cytometer was used to examine the cell apoptosis rate (Becton Dickinson, USA).
Statistical analysis
The mean standard deviation was utilized when the data were normally distributed and the independent t-test was used to compare the means of two independent groups. A p-value of less than 0.05 was considered statistically significant. Graphpad Prism 7.0 was used to conduct the statistical analyses (GraphPad Software Inc., USA).
Results
Histopathological changes in CIH-treated lung tissue of rats
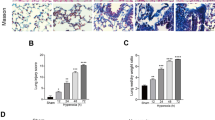
To observe whether CIH injured the lung architecture, HE staining and masson staining histopathological analyses of the lung tissue were performed. The lung tissue in the CIH group exhibited elevated lymphocyte and granulocyte infiltration compared with the control group. Furthermore, the CIH group had increased bleeding and edema in the alveolar compartment and increased thickness of the alveolar wall and vascular wall (Fig. 1A). Lung injury scores of CIH rats were significantly higher than those of control rats (p < 0.05) (Fig. 1B). Masson staining showed that CIH treatment increased the ratio of collagen (both p < 0.05) (Fig. 1C, D).

Histopathological alterations in lung tissue of rats after CIH treatment (n = 6). A HE staining of lung tissue in control group and CIH group; B Quantitative analysis of lung injury score based on HE staining; C Masson staining of lung tissue in control group and CIH group; D The proportion of collagen areas in lung tissue based on masson staining in control group and CIH group. Data are shown as the mean ± SD. ***p < 0. 001; ****p < 0. 0001
Ferroptosis in CIH-treated lung tissue of rats
The expression levels of GPX4 and ACSL4 were evaluated to verify ferroptosis in our study. Compared with the control group, ACSL4 mRNA levels were significantly up-regulated (Fig. 2A), while GPX4 mRNA levels were significantly down-regulated in the CIH group (Fig. 2B) (both p < 0.05). Simultaneously, ACSL4 protein levels were up-regulated and GPX4 protein levels were down-regulated in the CIH group (both p < 0.05), as shown in Fig. 2C.

The mRNA and protein levels of GPX4 and ACSL4 in CIH-treated lung tissue of rats (n = 6). A CIH increased the mRNA levels of ACSL4; B, CIH decreased the mRNA levels of GPX4; C The protein levels of ACSL4 and GPX4 in lung tissue of CIH-treated rats. Data are shown as the mean ± SD. **p < 0. 01; ***p < 0. 001
CIH-induced BEAS-2B injury and ferroptosis
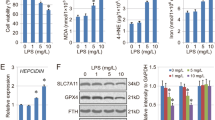
The cell viability of BEAS-2B reduced significantly after 24 h of intermittent hypoxia compared with the control group (100% ± 0% vs. 87% ± 4%, p < 0.05) (Fig. 3C). In the CIH group, the apoptotic rate of BEAS-2B increased greatly when compared with the control group (33.9% ± 1. 2% vs. 42.7% ± 0.3%, p < 0.05) (Fig. 3A). The levels of ROS were significantly elevated in the CIH group compared with the control group (p < 0.05) (Fig. 3B). The levels of IL-6 and TNFα mRNA were increased in CIH group compared with control group (both p < 0.05) (Fig. 3D). The mRNA and protein levels of ACSL4 were significantly higher in the CIH group than in the control group, while the mRNA and protein levels of GPX4 were significantly lower, all p < 0.05, as shown in Fig. 3E–G.

CIH-induced BEAS-2B injury and ferroptosis (n = 3). A CIH increased the apoptosis rate of BEAS-2B; B CIH increased the ROS levels of BEAS-2B; C CIH reduced cell viability of BEAS-2B; D CIH increased the mRNA levels of IL-6 and TNFα; E the mRNA levels of ACSL4 in Control group and CIH group; F the mRNA levels of GPX4 in Control group and CIH group; G The protein levels of ACSL4 and GPX4 in Control group and CIH group. Data are shown as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Fer-1 alleviated CIH-related BEAS-2B injury and ferroptosis
Cell viability improved (Fig. 4C), apoptosis rate decreased (Fig. 4A) and ROS accumulation decreased (Fig. 4B) in BEAS-2B co-treated with CIH and Fer-1 compared with the CIH group (all p < 0.05). In the CIH + Fer-1 group, the protein levels of ACSL4 decreased and the protein levels of GPX4 increased compared with the CIH group (both p < 0.05) (Fig. 4E), meanwhile the mRNA levels of IL-6 and TNFα decreased (both p < 0.05) (Fig. 4D).

Effects of Fer-1 on CIH-treated BEAS-2B (n = 3). A Fer-1 mitigated cellular apoptosis induced by CIH; B Fer-1 reversed the ROS accumulation induced by CIH; C Fer-1 improved the cell viability of CIH treated BEAS-2B; D Fer-1 reduced the mRNA expression levels of IL-6 and TNFα in CIH treated BEAS-2B; E the protein levels of GPX4 and ACSL4 in control group, CIH group and CIH + Fer-1 group of BEAS-2B. Data are shown as the mean ± SD. Compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; Compared with the CIH group, #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001
Fer-1 alleviated CIH related rats lung tissue injury and ferroptosis
We further conducted animal experiments to verify whether Fer-1 reversed CIH-induced lung injury. The results of HE staining showed that lung injury scores decreased after Fer-1 treatment (p < 0.05) (Fig. 5A, B). The results of masson staining showed that Fer-1 treatment attenuated the ratio of collagen caused by CIH (both p < 0.05) (Fig. 5C, D). The results of Western blot showed that Fer-1 treatment reversed the protein expression levels of ACSL4 and GPX4 (both p < 0.05) (Fig. 5E).

Effects of Fer-1 on CIH-treated lung tissue of rats (n = 6). A HE staining of lung tissue in control group, CIH group and CIH + Fer-1 group; B Quantitative analysis of lung injury scores based on HE staining; C Masson staining of lung tissue in control group, CIH group and CIH + Fer-1 group; D The proportion of collagen areas in lung tissue based on masson staining. E the protein levels of GPX4 and ACSL4 in control group, CIH group and CIH + Fer-1 group in lung tissue of rats. Data are shown as the mean ± SD. Compared with the control group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; Compared with the CIH group, #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001
Discussion
The current investigation demonstrated that CIH exposure increased the advancement of ferroptosis in bronchoalveolar epithelial cells and lung tissue. The intermittent hypoxia-related bronchoalveolar epithelial cell injury can be effectively inhibited by ferroptosis inhibitor Fer-1. This study provided a novel insight into CIH-induced lung injury.
Previous clinical studies found that OSA caused and aggravated various pulmonary diseases [20]. In addition, several prior animal investigations have verified lung injury caused by CIH [7, 21,22,23]. Inflammatory reactions and cellular apoptosis of lung tissue after intermittent hypoxia were the focus of previous studies [24, 25]. Ferroptosis is a novel type of cell death, which is different from apoptosis and necrosis [26]. Less attention has been focused on CIH-related ferroptosis in lung tissue. In this study, we established a CIH rat model to simulate the OSA hypoxic model. Besides histological lung tissue injury, we found ferroptosis-related protein dysregulation in the lung tissue of CIH-treated rats. Overexpression or knockdown of GPX4 regulated the cell mortality after various ferroptosis inducers treatment, so GPX4 has been identified as a key regulator of ferroptosis [27]. ACSL4 was identified as a key enzyme regulating lipid composition and promoting ferroptosis [28]. The decrease in GPX4 and the increase in ACSL4 have been utilized as indicators of ferroptosis. The dysregulation of GPX4 and ACSL4 in vitro and in vivo in this study confirmed the occurrence of ferroptosis after CIH treatment.
Ferroptosis was involved in the development and progression of a variety of lung diseases, including acute lung injury, COPD, and asthma [29,30,31]. Yoshida et al. [32] investigated the involvement of ferroptosis in the development and progression of COPD both in vitro and in vivo. Han et al. [33] found that IL-6 promoted ferroptosis in bronchial epithelial cells by inducing ROS-dependent lipid peroxidation. The injury induced by hypoxia and reoxygenation during CIH was similar to that induced by ischemia-reperfusion injury. Li et al. found that ferroptosis regulated intestinal ischemia/ reperfusion-induced acute lung injury via P53 [16]. We speculated the CIH-related ferroptosis in our study may also be associated with hypoxia and reoxygenation injury. Frequent cycles of hypoxia and reoxygenation resulted in the production of ROS, which triggered lipid peroxidation and ultimately ferroptosis [28]. In this study, the significant accumulation of ROS in CIH-treated BEAS-2B was consistent with our prediction. A graphic summary was shown in Fig. 6.

Proposed mechanisms of bronchoalveolar epithelial cells ferroptosis induced by CIH. CIH inhibited GPX4 activity which produced a lot of ROS, activated ACSL4, as a result promoted lipid peroxidation, and induced ferroptosis, eventually caused lung injury
One of the most well-known ferroptosis inhibitors is Fer-1 [34], which has been widely used in in vitro and in vivo studies. A previous study confirmed that Fer-1 alleviated cigarette smoke extract-induced BEAS-2B injury [35]. In addition, Fer-1 inhibited ferroptosis and alleviated lipopolysaccharide-induced acute lung injury [35]. Similar to previous studies, Fer-1 decreased the apoptosis rate and improved cell viability in CIH-treated BEAS-2B, and it also attenuated the pathological changes of lung tissue in CIH treated rats, which suggested the therapeutic potential of Fer-1 in CIH-related lung injury. Fer-1 usually ameliorated ferroptosis via inhibiting lipid peroxidation [36]. In the study, the ROS levels reduced after Fer-1 treatment, which confirmed Fer-1 alleviating oxidative stress in CIH-treated BEAS-2B. The mRNA levels of IL-6 and TNFα decreased after Fer-1 treatment, which indicated that Fer-1 reversed the inflammation caused by CIH exposure. In this study, the expression levels of GPX4 increased and the expression levels of ACSL4 decreased both in CIH-treated BEAS-2B and lung tissues of CIH-treated rats after treatment with Fer-1. These findings also showed Fer-1 reversed ferroptosis in CIH-treated BEAS-2B and lung tissue.
In addition to Fer-1, other types of ferroptosis inhibitors, lipoxstatin-1 and IASPP (an inhibitor of p53 transcriptional activity), were found to alleviate the ischemia-reperfusion-induced acute lung injury via inhibiting ferroptosis [37, 38]. The pathophysiological mechanisms of intermittent hypoxia and ischemia-reperfusion were similar. So the role of these ferroptosis inhibitors in CIH-related lung injury should be explored in future studies.
However, there are some limitations to this study. First, due to financial constraints, the sample size of animal experiments was relatively small. Second, although this study confirmed the occurrence of ferroptosis and the efficacy of Fer-1 in treatment of CIH-related lung injury, the mechanism was not explored in depth.
In conclusion, our results revealed that ferroptosis was involved in CIH-induced lung injury both in vitro and in vivo. Fer-1, a ferroptosis inhibitor, reversed the CIH-related lung injury.
Availability of data and materials
The data used in this study can be obtained through the corresponding author.
Abbreviations
- CIH:
-
Chronic intermittent hypoxia
- Fer-1:
-
Ferrostatin-1
- BEAS-2B:
-
Human bronchial epithelial cell line
- HE staining:
-
Hematoxylin–eosin staining
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- GPX4:
-
Glutathione peroxidase 4
- IL-6:
-
Interleukin-6
- TNFα:
-
Tumor necrosis factor α
- CCK-8:
-
Cell counting kit-8
- ROS:
-
Reactive oxygen species
- OSA:
-
Obstructive sleep apnea
- COPD:
-
Chronic obstructive pulmonary disease
- RIPA:
-
Radio immunoprecipitation assay
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TBST:
-
Tris buffer solution tween
- PBS:
-
Phosphate-buffered saline
References
Akashiba T, Inoue Y, Uchimura N, et al. Sleep apnea syndrome (SAS) clinical practice guidelines 2020. Respir Investig. 2022;60(1):3–32.
Yeghiazarians Y, Jneid H, Tietjens JR, et al. Obstructive sleep apnea and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;144(3):e56–67.
Yu Z, Cheng JX, Zhang D, et al. Association between obstructive sleep apnea and type 2 diabetes mellitus: a dose-response meta-analysis. Evid Based Complement Altern Med. 2021;2021:1337118.
Mcdermott M, Brown DL, Chervin RD. Sleep disorders and the risk of stroke. Expert Rev Neurother. 2018;18(7):523–31.
Dou L, Lan H, Reynolds DJ, et al. Association between obstructive sleep apnea and acute kidney injury in critically ill patients: a propensity-matched study. Nephron. 2017;135(2):137–46.
Bettini S, Serra R, Fabris R, et al. Association of obstructive sleep apnea with non-alcoholic fatty liver disease in patients with obesity: an observational study. Eat Weight Disord. 2022;27(1):335–43.
Lian N, Zhang S, Huang J, et al. Resveratrol attenuates intermittent hypoxia-induced lung injury by activating the Nrf2/ARE pathway. Lung. 2020;198(2):323–31.
Ding W, Zhang X, Zhang Q, et al. Adiponectin ameliorates lung injury induced by intermittent hypoxia through inhibition of ROS-associated pulmonary cell apoptosis. Sleep Breath. 2021;25(1):459–70.
Mankouski A, Kantores C, Wong MJ, et al. Intermittent hypoxia during recovery from neonatal hyperoxic lung injury causes long-term impairment of alveolar development: a new rat model of BPD. Am J Physiol Lung Cell Mol Physiol. 2017;312(2):L208–16.
Lin X, Jagadapillai R, Cai J, et al. Metallothionein induction attenuates the progression of lung injury in mice exposed to long-term intermittent hypoxia. Inflamm Res. 2020;69(1):15–26.
Emilsson ÖI, Sundbom F, Ljunggren M, et al. Association between lung function decline and obstructive sleep apnoea: the ALEC study. Sleep Breath. 2021;25(2):587–96.
Mcnicholas WT. COPD-OSA overlap syndrome: evolving evidence regarding epidemiology, clinical consequences, and management. Chest. 2017;152(6):1318–26.
Locke BW, Lee JJ, Sundar KM. OSA and chronic respiratory disease: mechanisms and epidemiology. Int J Environ Res Public Health. 2022;19(9):5473.
Naranjo M, Willes L, Prillaman BA, et al. Undiagnosed OSA may significantly affect outcomes in adults admitted for COPD in an Inner-City Hospital. Chest. 2020;158(3):1198–207.
Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30(6):478–90.
Li Y, Cao Y, Xiao J, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27(9):2635–50.
Chen LD, Wu RH, Huang YZ, et al. The role of ferroptosis in chronic intermittent hypoxia-induced liver injury in rats. Sleep Breath. 2020;24(4):1767–73.
Chen LD, Chen Q, Lin XJ, et al. Effect of chronic intermittent hypoxia on gene expression profiles of rat liver: a better understanding of OSA-related liver disease. Sleep Breath. 2020;24(2):761–70.
Fang YY, Luo M, Yue S, et al. 7,8-Dihydroxyflavone protects retinal ganglion cells against chronic intermittent hypoxia-induced oxidative stress damage via activation of the BDNF/TrkB signaling pathway. Sleep Breath. 2022;26(1):287–95.
Ioachimescu OC, Janocko NJ, Ciavatta MM, et al. Obstructive lung disease and obstructive sleep apnea (OLDOSA) cohort study: 10-year assessment. J Clin Sleep Med. 2020;16(2):267–77.
Braun RK, Broytman O, Braun FM, et al. Chronic intermittent hypoxia worsens bleomycin-induced lung fibrosis in rats. Respir Physiol Neurobiol. 2018;256:97–108.
Gille T, Didier M, Rotenberg C, et al. Intermittent hypoxia increases the severity of bleomycin-induced lung injury in mice. Oxid Med Cell Longev. 2018;2018:1240192.
Kang HH, Kim IK, Yeo CD, et al. The Effects of chronic intermittent hypoxia in bleomycin-induced lung injury on pulmonary fibrosis via regulating the NF-κB/Nrf2 signaling pathway. Tuberc Respir Dis (Seoul). 2020;83(Supple 1):S63-s74.
Gabryelska A, Łukasik ZM, Makowska JS, et al. Obstructive sleep apnea: from intermittent hypoxia to cardiovascular complications via blood platelets. Front Neurol. 2018;9:635.
Bikov A, Losonczy G, Kunos L. Role of lung volume and airway inflammation in obstructive sleep apnea. Respir Investig. 2017;55(6):326–33.
Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Lei G, Mao C, Yan Y, et al. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12(11):836–57.
Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–8.
Liu X, Zhang J, Xie W. The role of ferroptosis in acute lung injury. Mol Cell Biochem. 2022;477(5):1453–61.
Tang X, Li Z, Yu Z, et al. Effect of curcumin on lung epithelial injury and ferroptosis induced by cigarette smoke. Hum Exp Toxicol. 2021;40(12_suppl):S753–62.
Tang W, Dong M, Teng F, et al. Environmental allergens house dust mite-induced asthma is associated with ferroptosis in the lungs. Exp Ther Med. 2021;22(6):1483.
Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145.
Han F, Li S, Yang Y, et al. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered. 2021;12(1):5279–88.
Liu P, Feng Y, Li H, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 2020;25:10.
Lian N, Zhang Q, Chen J, et al. The role of ferroptosis in bronchoalveolar epithelial cell injury induced by cigarette smoke extract. Front Physiol. 2021;12:751206.
Shah R, Margison K, Pratt DA. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol. 2017;12(10):2538–45.
Yin X, Zhu G, Wang Q, et al. Ferroptosis, a new insight into acute lung injury. Front Pharmacol. 2021;12:709538.
Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88.
Acknowledgements
Not applicable.
Funding
This study was funded by National Natural Science Foundation of China (No. 82170101); Joint Funds for the Innovation of Science and Technology, Fujian Province (Grant number: 2019Y9116).
Author information
Authors and Affiliations
Contributions
NL designed and supervised the study. The first draft of the manuscript was written by JC, HZ and QC. Animal experiments, cell cultures, material preparation, data collection and analysis were performed by JC, YY, MC, MC and QC. JH and HZ participated in analysis of the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee of the Fujian Medical University Laboratory Animal Center approved this study (approve number: IACUCFJMU2022-0031). We verified that the study was reported following ARRIVE guidelines, all methods were performed in conformity with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
There is no conflict of interest between the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Original Western blot images in the manusctipt.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, J., Zhu, H., Chen, Q. et al. The role of ferroptosis in chronic intermittent hypoxia-induced lung injury. BMC Pulm Med 22, 488 (2022). https://doi.org/10.1186/s12890-022-02262-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-022-02262-x