
Abstract
Background
X-linked agammaglobulinemia (XLA) is an Inborn Errors of Immunity (IEI) characterized by pan-hypogammaglobulinemia and low numbers of B lymphocytes due to mutations in BTK gene. Usually, XLA patients are not susceptible to respiratory tract infections by viruses and do not present interstitial lung disease (ILD) such as bronchiolitis obliterans (BO) as a consequence of acute or chronic bacterial infections of the respiratory tract. Although many pathogenic variants have already been described in XLA, the heterogeneous clinical presentations in affected patients suggest a more complex genetic landscape underlying this disorder.
Case presentation
We report two pediatric cases from male siblings with X-Linked Agammaglobulinemia and bronchiolitis obliterans, a phenotype not often observed in XLA phenotype. The whole-exome sequencing (WES) analysis showed a rare hemizygous missense variant NM_000061.2(BTK):c.1751G>A(p.Gly584Glu) in BTK gene of both patients. We also identified a gain-of-function mutation in TGFβ1 (rs1800471) previously associated with transforming growth factor-beta1 production, fibrotic lung disease, and graft fibrosis after lung transplantation. TGFβ1 plays a key role in the regulation of immune processes and inflammatory response associated with pulmonary impairment.
Conclusions
Our report illustrates a possible role for WES in patients with known inborn errors of immunity, but uncommon clinical presentations, providing a personalized understanding of genetic basis, with possible implications in the identification of potential treatments, and prognosis for patients and their families.
Similar content being viewed by others

Background
X-Linked Agammaglobulinemia (XLA, OMIM entry 300,755) is a classic model of predominantly antibody deficiencies caused by mutations in Bruton’s Tyrosine Kinase (BTK) gene (OMIM #300300) with a reported incidence rate of 1/100,000 or 1/200,000 live births. The cytoplasmic tyrosine kinase protein encoded by BTK gene plays an important role in signal transduction during B cell maturation [1, 2]. Due to arrest of B cell development at the pre-B or mature B cells differentiation stages, affected patients usually display reduced total immunoglobulin (Ig) production and peripheral B cells frequency (CD19 and CD20) less than 1–2% [1, 3]. Male patients with XLA frequently undergo recurrent infection episodes such as otitis, pneumonia, sinusitis, and arthritis during childhood.
This increased risk for pulmonary complications, even after Ig replacement therapy (IRT), is often caused by bronchiectasis and interstitial lung disease (ILD) due to bacterial infections [4, 5]. However, ILD shows a significantly variable clinical manifestation among patients with primary antibody deficiency (PAD), suggesting the influence of antibody-independent factors not yet defined. Cases of ILD in XLA patients are atypical when compared to common variable immunodeficiency (CVID), and, overall, they are reported by multicenter studies focusing on general clinical and laboratory comparison rather than genetic and epigenetic findings [6]. Genetic contributions to the progression of lung complications in XLA regardless of IRT are still not elucidated.
Here, we report two pediatric cases from male siblings with severe B lymphopenia, recurrent infection episodes, and antibody deficiency diagnosed as XLA. Interestingly, both patients presented severe pulmonary impairment diagnosed as bronchiolitis obliterans using tomographic criteria, not often observed in XLA. We carried out a comprehensive whole-exome sequencing study in the subjects aiming to identify candidate genetic variants possibly related to the both clinical presentations observed. Variant prioritization was conducted using the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines for sequence variants analysis in order to provide a precise genetic diagnosis and better comprehend the genetic mechanisms underlying XLA in our patients.
Case presentation
Patient 1 (P1) and 2 (P2) are two male siblings from a non-consanguineous couple, born in the state of Rio de Janeiro, Brazil. They had a past clinical history of recurrent respiratory infections, mainly pneumonia, severe wheezing, and sepsis, with symptoms onset being observed before the first year of life in P1 and at 2 y.o for P2. Severe wheezing and secondary bacterial pneumonia were part of the initial presentation. Both children were first seen during their first ICU hospitalization. The XLA diagnosis was established at 11 months and 2y5m of age in each subject, respectively. At the time this study was conducted, P1 was 3 y.o and P2 was 5 y.o.
Immunological findings revealed CD19 and CD20 less than 1% in both patients. P1 showed levels of IgG < 270 mg/dL (Reference Value for Brazilian patients (RV), RV: 520–875 mg/dL), IgM = 12 mg/dL (RV: 47–138 mg/dL), and IgA = 20 mg/dL (RV: 7–130 mg/dL). On the other hand, P2 had IgG = 100 mg/dL (RV: 540–116 mg/dL), IgM = 24.9 mg/dL (RV: 43–194 mg/dL), IgA < 5 mg/dL (RV: 11–192 mg/dL). Both had undetectable levels of IgE isotype.
Patients were kept on the regular infusion of intravenous immunoglobulin replacement and prophylaxis with azithromycin after the diagnosis. They were receiving Immunoglobulin reposition doses (600 mg/kg every 21 days IV). We kept both on azithromycin as soon as they were discharged from ICU. They had already been diagnosed as probable XLA due to agammaglobulinemia and low B cells and presenting persistent wheezing.
Both patients had recurrent and severe wheezing and images suggesting bronchiolitis obliterans were observed in the thorax computed tomography (CT; Figs. 1 and S1). They evolved with recurrent respiratory viral infections, severe bronchospasm, and the need for several hospitalizations, as well as worsening of CT images. In both patients, no severe bacterial infections have been observed since the beginning of immunoglobulin replacement. Currently, patient 1 is 6 y.o and has partial control of the bronchospasm, even with high doses of inhaled corticosteroids, associated with long-acting beta-agonists (LABA) and long-acting muscarinic antagonist (LAMA). Now he presents signs of chronic pulmonary disease: increase in chest anteroposterior diameter and digital clubbing. Patient 2, evolved with more severe pulmonary disease and was kept also on high doses of inhaled corticosteroid, LABA and LAMA, plus oral cyclosporine and frequent oral corticosteroid short treatments. However, he died with respiratory insufficiency at 6 y.o.

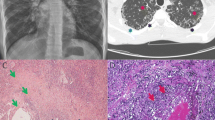
Thorax CT imaging of the two study subjects. Upper and lower rows indicate patient 1 (a-c) and patient 2 (d-f) images, respectively. a-c) was obtained in 2016 at 3 y.o of P1. Chest CT scan in d) was obtained in 2015 at 3y7mo, whereas e and f were taken in late 2017 at 6 y.o. Red arrows indicate mosaic attenuation patterns and Bronchiectasis. Yellow arrow shows atelectasis. In pink, we showed a large bronchiectasis and bronchial wall thickening
On average, whole-exome sequencing (WES) achieved 99% of the reads mapped to the human reference genome with the mean depth of coverage greater than 100X in 96% of exonic regions. A total of 73,668 SNVs were identified among common and rare variants, being 40,372 shared in both patients. Due to the potential presence of non-synonymous variants in genes previously associated with inborn errors of immunity (IEI), we queried an enriched set of genes related to the agammaglobulinemia phenotype (HP:0,004,432) according to HPO [7]. From the 19 genes filtered, we only found variants in the BTK gene of both patients. The variant was located in the exon 16 of the NM_000061.2 transcript, the candidate to be the principal isoform of the gene (Figure S2). The missense variant changes a nonpolar residue of Glycine at the codon 584 by a polar neutrally charged Glutamate in the TK domain of the BTK protein. NM_000061.2(BTK):c.1751G>A (p.Gly584Glu) was not found in control samples from the 1000 Genomes Project and the Genome Aggregation Database (GnomAD). This variant is located on the exonic side of the splice site with a predicted loss-of-function (pLOF) effect (gene has 121 pathogenic LOF variants and gnomAD Loss-of-Function Observed/Expected = 0 is less than 0.755). The codon 584 is located in a mutational hot-spot with 17 amino acids harboring 7 non-VUS (7 pathogenic and 0 benign). Missense variants are a common driven mechanism of XLA disease in BTK. We also observed 12 pathogenic predictions from BayesDel_addAF, CADD, DEOGEN2, FATHMM-MKL, LIST-S2, M-CAP, MVP, MutationAssessor, MutationTaster, Polyphen2-HVAR, PrimateAI, and SIFT versus no benign predictions. This variant was also reported as pathogenic in the literature in patients with XLA [8, 9]. Altogether, Gly584Glu was classified as Pathogenic by Varsome using the following ACMG/AMP criteria: PVS1, PM1, PM2, PP2, PP3.
Next, we retrieved the HPO terms for bronchiolitis obliterans (HP:0,011,946), bronchiolitis (HP:0,011,950), bronchospasm (HP:0,025,428) and pneumonia (HP:0,002,090) phenotypes in HPO database. An additional list of genes associated with bronchiolitis was also retrieved from the Human Gene Mutation Database (HGMD; see supplementary material). We found a missense variant (rs1800471) in the TGFβ1 gene reported as a disease-associated polymorphism with supporting functional evidence in HGMD. The Arg25Pro has been associated with transforming growth factor-beta1 production, fibrotic lung disease, and graft fibrosis after lung transplantation.
We also predicted the classic HLA alleles [HLA-A, HLA-B, HLA-C, HLA-DP (DPA1, DPB1, and DPB2), HLA-DQ (DQA1, DQA2, and DQB1), and HLA-DR (DRA, DRB1, 3, 4, and 5)] for each patient using NGS data (Table S1). Patient 1 carried the haplotype DRB3*02:02 associated with bronchospasm [10]. Three other haplotypes DRB1*11:01, DQB1*03:01, and DQA1*05 were found in this subject previously associated with sepsis and pneumonia [11,12,13]. HLA alleles associated with symptoms of bronchospasm were also found in Patient 2 (DPB1*03:01), which also harbored DQB1*05:02 and DRB1*16:02 often found in patients with sepsis [14, 15].
Discussion and conclusions
The patients reported here were diagnosed with XLA after an initial episode of probably bacterial lung infection associated with sepsis and severe bronchospasm. Since the beginning of regular replacement of intravenous immunoglobulin and initial use of prophylactic antibiotics, they have had no relevant bacterial infection. However, the clinical course of both was characterized by recurrent and severe episodes of wheezing, many of them requiring hospitalization and insufficient response to the proposed treatment. The clinical and radiological images of both patients were compatible with the diagnosis of post-infectious bronchiolitis obliterans. Post-infectious bronchiolitis obliterans occurs most commonly after viral respiratory tract infections by adenovirus, or more rarely related to bacterial infections [16]. Overall, patients with XLA are not susceptible to respiratory tract infections by viruses and also do not usually present BO as a consequence of acute or chronic bacterial infections of the respiratory tract [6, 17].
Bronchiolitis obliterans (BO) is an ILD characterized by subepithelial inflammation and airway obstruction due to fibrosis of the bronchioles, which leads to defects in epithelial and airway regeneration. BO is often associated with lung or hematopoietic stem cell transplants, autoimmune disorders, inhalation of toxic agents, and post-infectious episodes. Barnes et al. 2015 reported a series of XLA patients who developed BO after lung transplantation [18]. Adenovirus infection is among the main potential causes of BO from post-infectious origins [19, 20]. Weinberger et al. 2019 estimated a rare incidence of less than 1% of BO manifestation in XLA patients from the US Immunodeficiency Network [21]. For this reason, the mechanisms underlying the risks of developing BO in XLA are still poorly understood. Even with the use of regular IRT with proper dosage, infections on the surface of the respiratory tract may continue to occur. This constitutes an important factor for the development of bronchiectasis and chronic lung disease, which is usually proportional to the years of the disease.
In the present study, we detected a heterozygous gain-of-function (GoF) variant in TGFβ1 (rs1800471) in two siblings with clinical manifestations of X-Linked Agammaglobulinemia. Although the rs1800471 seems to play a role in the phenotype observed in our patients, the limited number of samples does not allow us to draw conclusions about its association with the phenotype of bronchiolitis obliterans. Further studies are needed to confirm this association in other patients with IEI, particularly those with defects in antibody production, and the diagnosis of post-infectious bronchiolitis obliterans. The haplotype of classic HLA alleles may also contribute to the phenotype observed once similar related symptoms were described in the literature in other individuals. Both HLA alleles and TGFβ1 variant seem not to have a causative effect, instead, we hypothesized that they might be associated with the phenotypes only based on the evidence available in the literature. Therefore, patients were diagnosed with XLA and may have modifiers genes. Altogether, our report illustrates a possible role for WES in patients with known IEI and uncommon clinical presentations, providing a personalized understanding of genetic basis, with possible implications in the identification of potential treatments, and prognosis for patients and their families.
Availability of data and materials
The sequencing data used in our study are publicly available in SRA-NCBI (www.ncbi.nlm.nih.gov/sra), SRA accession: PRJNA818322.
Abbreviations
- XLA:
-
X-linked agammaglobulinemia
- IEI:
-
Inborn errors of immunity
- ILD:
-
Interstitial lung disease
- BO:
-
Bronchiolitis obliterans
- WES:
-
Whole-exome sequencing
- BTK:
-
Bruton’s Tyrosine Kinase
- PAD:
-
Primary antibody deficiency
- CVID:
-
Common variable immunodeficiency
- CD19:
-
Cluster of Differentiation 19
- CD20:
-
Cluster of Differentiation 20
- Ig:
-
Immunoglobulin
- IRT:
-
Immunoglobulin replacement therapy
- AMP:
-
Association for Molecular Pathology
- ACMG:
-
American College of Medical Genetics and Genomics
- CT:
-
Computer tomography
- LABA:
-
Long-acting beta-agonists
- LAMA:
-
Long-acting muscarinic antagonist
References
Suri D, Rawat A, Singh S. X-linked Agammaglobulinemia. Indian J Pediatr. 2016;83:331–7.
Smith CI, Baskin B, Humire-Greiff P, Zhou JN, Olsson PG, Maniar HS, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152:557–65.
Smith T, Cunningham-Rundles C. Primary B-cell immunodeficiencies. Hum Immunol. 2018. https://doi.org/10.1016/j.humimm.2018.10.015.
Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109:1001–4.
Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: A meta-analysis of clinical studies. Clin Immunol. 2010;137:21–30.
Pac M, Dmenska H, Bernatowska E. Pulmonary manifestation of X-linked agammaglobulinemia. Eur Respir J. 2014;44(Suppl):58.
Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2020;49:D1207–17.
Velickovic M, Prasad ML, Weston SA, Benson EM. Identification of the bruton tyrosine kinase (BTK) gene mutations in 20 Australian families with X-linked agammaglobulinemia (XLA). Hum Mutat. 2004;23:398–9.
Domingo A, Schmidt TGPM, Barcelon E, Lukban M, Westenberger A, Klein C. X-linked agammaglobulinemia with hearing impairment, dystonia-parkinsonism, and progressive neurodegeneration. J Neurol. 2014;261:2225–7.
Romano A, Oussalah A, Chery C, Rodriguez-Gueant RM, Gaeta F, Cornejo-García JA, et al. Next-Generation Sequencing and Genotype Association Studies Reveal the Association of HLA-DRB3*02:02 With Delayed Hypersensitivity to Penicillins. Authorea Preprints. 2021. https://doi.org/10.22541/au.161857346.60929786/v1.
Chi C-Y, Chu C-C, Liu J-P, Lin C-H, Ho M-W, Lo W-J, et al. Anti–IFN-γ autoantibodies in adults with disseminated nontuberculous mycobacterial infections are associated with HLA-DRB1*16:02 and HLA-DQB1*05:02 and the reactivation of latent varicella-zoster virus infection. Blood. 2013;121:1357–66.
Eglite E, Hagina E, Pavare J, Grope I, Eihvalde L, Sochnevs A, et al. Genetic Polymorphisms HLA Class II in SIRS and Sepsis in Children. J Adv Med Med Res. 2014;4:149–60.
Bridina L, Silina S, Eglite J, Krumina A. Determination of HLA - DRB1, -DQA1 and -DQB1 alleles frequency in sepsis patients. Int J Infect Dis. 2018;73:196–7.
Dekker JW, Nizankowska E, Schmitz-Schumann M, Pile K, Bochenek G, Dyczek A, et al. Aspirin-induced asthma and HLA-DRB1 and HLA-DPB1 genotypes. Clin Exp Allergy. 1997;27:574–7.
Suárez I, Lehmann C, Gruell H, Graeb J, Kochanek M, Fätkenheuer G, et al. Repurposing QuantiFERON for Detection of Neutralizing Interferon-γ Autoantibodies in Patients With Nontuberculous Mycobacterial Infections. Clin Infect Dis. 2017;65:518–21.
Zhang L, Silva FA e. Bronchiolitis obliterans in children. J Pediatr (Rio J). 2000;76:185–92.
Plebani A, Lougaris V, editors. Agammaglobulinemia. 1st ed. Cham, Switzerland: Springer International Publishing; 2015.
Barnes S, Kotecha S, Douglass JA, Paul E, Hore-Lacy F, Stirling R, et al. Evolving practice: X-linked agammaglobulinemia and lung transplantation. Am J Transplant. 2015;15:1110–3.
Lynch JP 3rd, Weigt SS, DerHovanessian A, Fishbein MC, Gutierrez A, Belperio JA. Obliterative (constrictive) bronchiolitis. Semin Respir Crit Care Med. 2012;33:509–32.
Li Y-N, Liu L, Qiao H-M, Cheng H, Cheng H-J. Post-infectious bronchiolitis obliterans in children: a review of 42 cases. BMC Pediatr. 2014;14:238.
Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol. 2019;39:440–7.
Acknowledgements
We thank the patients and their families for taking part in this study.
Funding
This research was supported by grants from the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ #E-26/202.826/2018 (BR) and # E-26/210.086/2022 (BR), Conselho Nacional de Desenvolvimento Científico e Tecnológico–CNPq # 303170/2017–4 (BR), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) [Grant: Biocomputacional # 23038.010041/2013¬13], RSFJ received graduate fellowships from the Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–CAPES. The funding body had no role in the design of the study and collection, analysis, and interpretation of data, or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ATRV, ZFMV, ESG, FPM and FAA conceived and designed the project and are responsible for the overall content. RSFJ, GLM, JBC, and CSF performed the bioinformatics analysis of WES data. ALG, and APCG performed sequencing experiments. ESG and FPM collected the clinical data. RSFJ, GLM, JBC, FPM, FAA, ESG, ZFMV, and ATRV prepared the manuscript. All authors have read, revised, and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Ethical Committee of the Instituto Fernandes Figueira (no. CAAE42934815.4.0000.52695269) and by the Ethical Committee of the Instituto Nacional do Cancer (153/10).
Consent for publication
Written informed consent was obtained for both patients from their guardians to publish the children's personal and clinical details along with any accompanying data used in this study.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
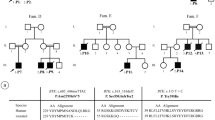
Table S1. Prediction of HLA alleles present in each patient. Figure S1. Chest CT scan of patient 2. a-c) was obtained in 2015 at 3y7mo, whereas d was obtained in late 2017 at 6 y.o. Cyan arrows show bronchial wall thickening. Red arrows indicate mosaic attenuation patterns and Bronchiectasis. Yellow arrow shows atelectasis. Figure S2. Genetic diagnosis of XLA patients using WES. a) Ideogram of the human X chromosome, in red the region q22.1 where the BTK gene is located, and the BTK isoform taken from UCSC Genome Browser. b) Family pedigree showing the inheritance model of BTK mutation in both siblings. c) The box represents a zoom in the region where the mutation is located. Red and blue rectangles represent the forward and reverse NGS reads covering the variants, followed by the Sanger sequencing eletrofluorograms for the same region.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Francisco Junior, R., de Morais, G.L., de Carvalho, J.B. et al. Clinical and genetic findings in two siblings with X-Linked agammaglobulinemia and bronchiolitis obliterans: a case report. BMC Pediatr 22, 181 (2022). https://doi.org/10.1186/s12887-022-03245-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-022-03245-x