
Abstract
Background
Sandhoff disease (SD) is an autosomal recessive lysosomal storage disorder, resulting in accumulation of GM2 ganglioside, particular in neuronal cells. The disorder is caused by deficiency of β-hexosaminidase B (HEX-B), due to pathogenic variant of human HEXB gene.
Method
This study describes clinical features, biochemical, and genetic defects among Thai patients with infantile SD during 2008–2019.
Results
Five unrelated Thai patients presenting with developmental regression, axial hypotonia, seizures, exaggerated startle response to noise, and macular cherry red spot were confirmed to have infantile SD based on deficient HEX enzyme activities and biallelic variants of the HEXB gene. In addition, an uncommon presenting feature, cardiac defect, was observed in one patient. All the patients died in their early childhood. Plasma total HEX and HEX-B activities were severely deficient. Sequencing analysis of HEXB gene identified two variants including c.1652G>A (p.Cys551Tyr) and a novel variant of c.761T>C (p.Leu254Ser), in 90 and 10% of the mutant alleles found, respectively. The results from in silico analysis using multiple bioinformatics tools were in agreement that the p.Cys551Tyr and the p.Leu254Ser are likely pathogenic variants. Molecular modelling suggested that the Cys551Tyr disrupt disulfide bond, leading to protein destabilization while the Leu254Ser resulted in change of secondary structure from helix to coil and disturbing conformation of the active site of the enzyme. Genome-wide SNP array analysis showed no significant relatedness between the five affected individuals. These two variants were not present in control individuals. The prevalence of infantile SD in Thai population is estimated 1 in 1,458,521 and carrier frequency at 1 in 604.
Conclusion
The study suggests that SD likely represents the most common subtype of rare infantile GM2 gangliosidosis identified among Thai patients. We firstly described a potential common variant in HEXB in Thai patients with infantile onset SD. The data can aid a rapid molecular confirmation of infantile SD starting with the hotspot variant and the use of expanded carrier testing.
Similar content being viewed by others


Background
GM2 gangliosidoses including Tay-Sachs disease (TSD, MIM 272800), Sandhoff disease (SD, MIM 268800), and GM2 activator protein deficiency (GM2AP or AB variant, MIM 272750) are a group of lysosomal storage disorders of which the breakdown of glycosphingolipids, GM2 gangliosides, is impaired, leading to an accumulation of the substrates in various internal organs, particular in neuronal cells [1]. These disorders are resulted from deficiency of β-hexosaminidase enzyme (HEX), or rarely, defect of activator protein. Two isoforms of HEX includes β-hexosaminidase A (HEX-A) consisting of a heterodimer between α- and β-subunit (αβ), and β-hexosaminidase B (HEX-B) consisting of a homodimer of β-subunit (ββ) [2]. TSD, SD, and GM2AP deficiency are caused by biallelic pathogenic variants of human HEXA, HEXB, GM2AP gene in respective order [1]. HEXB is located on 5q13, containing 14 coding exons and spanning mRNA of 2 kb, and encoding 556 amino acids.
There are three clinical subtypes in TSD and SD including infantile, juvenile, and adult subtypes [1]. Infantile manifestations of TSD, SD, and GM2AP are clinically indistinguishable. The infantile disease is characterized by development of axial hypotonia, startle response with hyperacusis, developmental regression starting at the age of 3–6 months, followed by progressive loss of motor skill, neurologic signs of upper and lower motor neurons, macrocephaly, seizures, macular cherry-red spots, and eventually death before 3–5 years of age [1, 3, 4].
Patients with juvenile and adult forms present in early-late childhood or adulthood with slow progression of neurological symptoms such as developmental delay, abnormal gait, ataxia, spinocerebellar and motor neuron dysfunction, increased myotatic reflexes which sometimes are misdiagnosed with late-onset spinal muscular atrophy, and Friedreich ataxia [1, 5].
The diagnosis of SD can be confirmed by biochemical findings of low total HEX and deficient HEX-B activities, with high percentage of HEX-A/total HEX activity.
The prevalence of GM2 gangliosidosis in Thailand is not known. Only two cases of SD without molecular data [6, 7] and a single patient with TSD [8] were previously described. During a 12-year period of 2008–2019, our laboratory is only one center in Thailand to provide biochemical analysis for GM2 gangliosidoses, of which 5 out of 8 specimens were confirmed to have SD and none for TSD. Herein, we describe clinical, biochemical, and molecular characteristics of the five patients with infantile-onset SD and HEXB variants found among the Thai patients.
Methods
Patients
Five unrelated patients (2 female and 3 male) biochemically confirmed of having SD were included in the study. Genetic analysis was performed following the approval of Ramathibodi Hospital Institutional Review Board.
Biochemical analysis
Measurement of the patients’ plasma total HEX and HEX-B enzyme activity was determined using fluorogenic substrate (0.95 mg/ml) 4-methylumbelliferyl-2-acetomido-2-deoxy-β-D-glucopyranoside (4MUG, Sigma, MO) with differential heat inactivation, following established protocol [9]. Briefly, total HEX activity was estimated by incubation at 37 °C for 20 min in the presence of 2.5 mM 4MUG and 0.01 M citrate-phosphate buffer, pH 4.4 in a total volume of 0.25 ml [9]. Heat inactivation was processed by pre-incubating samples for 180 min at 52 °C before adding 4MUG and incubation at 37 °C for 20 min; after which reactions were stopped by adding 1.25 ml of 0.17 M glycine-carbonate buffer, pH 9.8. This process led to loss of HEX-A, which is heat-labile [9]. Therefore, HEX-B was determined as the activity at this point. Fluorescence excitation was processed at 365 nm wavelength and emission was determined at 450 nm. The %HEX-A activities were calculated by subtracting HEX-B activity from total HEX activity. All samples were performed in triplicate.
Genetic analysis
Genomic DNA was extracted from peripheral blood specimens following standard protocols (QIAGEN GmbH). PCR primers for each of the coding exons of HEXB gene and its intron-flanking sequences were designed using program PRIMER 3 (http://www.frodo.wi.mit.edu/cgi-bin/primer3). GenBank reference sequences were NT_006713, NM_000521 and NP_000512. PCR and direct sequencing of all the exons were performed, primer sequences and PCR conditions were provided as supplemental data (Table S1).
Once a variant(s) was identified, specimens from parents and available sibs were tested for the variant discovered. For screening of variants identified, 50 healthy control individuals (regular blood donors) were analyzed using restriction digest with appropriate endonuclease restriction enzyme(s). Later, data from an in-house (unpublished) whole exome database of 496 Thai individuals affected with other known/unknown disorders but not related to the SD-related phenotypes was checked for the presence of the mutant allele. To predict the effect of missense variant, we performed in silico analysis using multiple software tools. Multiple protein sequence alignment of vertebrate species was performed by using Clustal Omega software (https://www.ebi.ac.uk/Tools/msa/clustalo).
Prediction of the structure and function of the mutant protein
To predict the structural and functional changes of the HEXB mutant protein, I-TASSER, COTH and Mutation Taster were used for analysis (reference sequence NP_000512.1) [10,11,12]. The 3D homological structures of mutant protein were constructed using SWISS-MODEL [13]. The HEXB with PDB-ID: 1o7a was choose as the template [14]. The model was visualized, compared and analyzed using UCSF chimera [15].
Detection of common founders
To identify common ancestry among the patients with the rare variant detected, we performed whole genome genotyping using Infinium Asian Screening Array version 1.0. All five probands were genotyped including 19 randomly selected unrelated control from Thai population.
A total of 35,725 SNPs on chromosome 5 were converted to forward strand base and non-polymorphic variants were removed, leaving 25,624 SNPs for subsequent analysis.
Phasing was performed using Eagle version 2 [16] with the non-European or mixed population from the Haplotype Reference Consortium (HRC Version r1.1 2016) [17] as a reference population. Phasing was carried out on the Michigan Imputation Server [18].
To detect a shared identity-by-descent/identity-by-state (IBD/IBS) indicating shared founders among cases, we used GERMLINE version 1.5.3 [19] specifying the option -min_m 1 and -bits 50 aiming to detect a minimum size of 1 cM IBD segment. A total of 18,733 polymorphic SNPs with minor allele frequency (MAF) > 0.05 were included in this part of the analysis. Subsequently, ERSA version 2.1 was used to test for a statistically significant shared recent common ancestry [20].
Results
Clinical characteristics
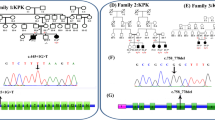
All patients were unrelated and lived in different provinces located in the Central (1), Northern (1) and Northeastern (3) Thailand. The mean age of disease onset was 6.6 months and the median age of diagnosis was 17 months (range 16–34 months) (Table 1). The mean duration of time to diagnosis was 15 months (range 11–26 months). Developmental regression is the first manifestation in all patients. Despite normal psychomotor development during the first 6 months of life, patient-1 had been misdiagnosed of having cerebral palsy. At the age of 9 months, patient-1 was incidentally heard to have heart murmur and later confirmed to have mild mitral valve prolapse and moderate mitral regurgitation [6]. Macular cherry red spot and excessive startle response to noise were present in all patients. Macrocephaly was noted in two, and seizures in all of the patients. Myoclonic seizure exacerbated by loud noise was observed in 3 out of 5 patients whereas generalized tonic clonic seizures were also commonly found (Table 1). Mild prematurity was observed in two patients. Four patients were sporadic cases, except for patient-1 who had a deceased elder brother who died due to progressive neurological regression of unclear etiology at the age of 3 years (Fig. 1a). Parental consanguinity was present in family-2.

Pedigrees of 5 affected families. Noted their genotypes, black square/circle indicating affected individual
Enzyme activities and HEXB variants
All patients have significantly low total HEX activities ranging from 3.0–9.6% of normal control. The HEX-B activities were ranging 0.9–2.7% of normal control, while % of HEX-A/total HEX activities were elevated as expected (Table 2).
A nucleotide substitution, c.1652G>A (p.Cys551Tyr) of the exon 14 was identified in homozygous state in 4 patients and present in compound heterozygous state with another variant (in trans), c.761T>C (p.Leu254Ser) of exon 6, in one patient (Fig. 2a). Eight of 10 parents were tested and found to be heterozygous for either p.Cys551Tyr or p.Leu254Ser. The in silico analyses using SIFT, PolyPhen2, PROVEAN, PredictSNP2, CADD, DANN, FATHMM, FunSeq2 and GWAVA were in agreement, suggesting deleterious effect of both missense alleles (supplemental data, Table S2). The c.1652G>A variant created a PsiI restriction site which was used as the second method to confirm the presence of this variant in the patients, relatives and controls (Fig. 2b). Comparative in silico analysis of the p.Cys551Tyr of and the p.Leu254Ser showed evolutionary conservation among vertebrate species of Cys551 and Leu254 residues (Fig. 2c). The two variants were not found in our in-house whole exome database and in the gnomAD (allele frequency = 0). The c.1652G>A was assigned as uncertain significance (PM2, PP2, PP3) while the c.761T>C was classified as likely pathogenic (PM2, PP2, PP3, PP5), based on American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (https://varsome.com/variant/hg19).

Mutant sequences and protein alignment. a Genomic DNA sequences showing HEXB variants. Noted heterozygous c.761T>C (p.Leu254Ser) in patient-4, homozygous c.1652G>A (p.Cys551Tyr) in patient-1, and sequence of normal control on side by side. b PCR-PsiI restriction digest. Noted the c.1652G>A mutant allele creating a Psil restriction site, yielding 179 and 191 bp allele in homozygous individuals (patient-1, −2 and − 3 in lanes 2, 4 and 6, respectively) whereas heterozygous individuals harbored the uncut normal allele of 370 bp in addition to the 179 and 191 bp from the mutant allele (unaffected brother of patient-1 and the mother of patient-2 in lanes 3 and 5, respectively). c Protein sequence alignment of HEXB across various vertebrate species. Noted highly conserved nature of the Cysteine551 and the Leucine254 residues
Molecular modelling of the mutant protein
The overall features of structural analysis were summarized in Table 3. The homological structures were able to build with qualitative model energy analysis (QMEAN) value indicating that the predicted structure was with good quality [21]. The replacement of Cys551 by Tyr led to the loss of disulfide bond with Cys534 and created a hydrogen bond formation with the carboxyl group of Thr530 as predicted by MutationTaster (Fig. 3a). The variant from Leu254 to Ser254 resulted in altered secondary structure, from helix (amino acid 253–258) to coil as indicated by I-TASSER and MutationTaster. The p.Leu254Ser created one additional hydrogen bond formed between the hydroxyl group of Ser254 and the carboxyl group of Phe251, which is predicted to destroy the helical structure and decrease protein stability (Fig. 3b) [22].

Structural homology of the Cys551Tyr and Leu254Ser. a Molecular model of Cys551Tyr. Noted loss of the disulfide bond between Cys534 and Cys551 and additional hydrogen bond formation between Cys534 and Thr530. b Homological model of Leu254Ser, indicating one additional hydrogen bond forming between Ser254 and Phe251
COTH and SWISS-MODEL showed that homodimerization Cys551Tyr-Cys551Tyr could be formed in the case of homozygous state whereas Cys551Tyr-Cys551Tyr, Leu254Ser-Leu254Ser, and Leu254Ser-Cys551Tyr could be found in compound heterozygosity, though the dimers may be less stable in both cases. SWISS-MODEL was able to predict the homodimerization between the two identical subunits, while COTH was more flexible to predict the homodimerization of the non-identical subunits (Table 3). The quaternary structure quality estimate (QSQE) is equal to 1 (range 0–1), reflecting accurate prediction of the oligomerization [23].
Common founders
GERMLINE identified 1539 segments shared IBS with the median size of 1.43 cM (range from 1 to 4.58). On average, the combined shared segments between each individual was 9.39 cM (SD = 0.519). However, there was no significant relatedness detected between each pair of cases (p-value = 1).
Discussion
We described five patients with infantile onset SD who presented with characteristic features of GM2 gangliosidosis. Pattern of generalized tonic-clonic seizures and myoclonic seizures exacerbated by loud noise was consistent with previous observation [3]. Unlike TSD, mild hepatosplenomegaly can be noted in SD as also found in one patient of the present cohort (patient-3) [24]. Cardiac involvement including cardiomegaly, valvulopathies, hypertrophic/dilated cardiomyopathy is an uncommon finding, it is rare but could be a presenting feature of infantile SD and even ahead of neurological manifestation [25, 26].
Late stage SD could be misdiagnosed as cerebral palsy and the cause of seizures was though due to cerebral palsy and vice versa, as seen in one of our patients. A careful history taking including detailed progression of psychomotor retardation and seizures pattern is essential to alert pediatricians to this rare disorder.
For the first time, we described molecular characteristics of infantile SD in Southeast Asian descendants which is an underrepresented population. It is surprising that most of the Thai patients shared a common allele, c.1652G>A (p.Cys551Tyr). The mutant p.Cys551Tyr is expected to cause protein destabilization due to disruption of its disulfide bond formed with the Cys534. Amino acid residue 547–552 are involved in C-terminal looping which is part of the dimerization; therefore, Cys551Tyr could compromised the dimer formation, and disabled the active site of the protein [14]. The additional hydrogen bond formed between Tyr551 and Thr530 can affect the dipole moment of α-helix.
The novel c.761T>C (p.Leu254Ser; ClinVar# SCV000999190) allele was found in compound (trans) with the c.1652G>A variant in a SD patient (patient-4) whose parents were heterozygous for one of the variants. The substitution of leucine residue, a non-polar aliphatic amino acid to serine which is a polar amino acid, likely leads to altered secondary/tertiary structure of the protein and hence deleterious effect. The active sites of HEX-B enzyme compose of Arg211, Asp240, His294, Asp354, Glu355, Tyr450, Asp452, and Glu491 (Fig. 3b) which are located at (β/α)8-barrel structure [14]. The Leu254 is surrounded by bulky hydrophobic amino acids (Phe244, Phe246, Phe251, Trp298, Phe332, Phe336, and Phe337) and is part of the helical extension (252–259) of (β/α)8-barrel structure [14]. The replacement of Leu254, a hydrophobic amino acid by a more hydrophilic amino acid, Ser, could interfere the hydrophobic intermolecular contacts, leading to disruption of the cavity of the enzyme active site (Fig. 3b). Enzyme kinetic experiment or in vitro functional study is needed to confirm the pathogenic molecular mechanism of the Cys551Tyr and Leu254Ser.
The variant c.1652G>A or p.Cys551Tyr (ClinVar# VCV000167176) was first reported without clinical data in a study of SD carrier frequency for Saskatchewan in Canada of which a combined HEX-B enzyme activity and variant analysis identified a case of SD with genetic compounds between c.1652G>A and c.115delG [27]. Recently, the c.1652G>A was again described in compound heterozygous state with a nonsense variant p.Tyr463* in a Chinese infant with SD [28]. Taken together with being the common variant shared among the Thai affected infants, its absence in our control population, the highly conserved codon of Cys551, and the molecular modelling, the c.1652G>A is most likely a pathogenic allele.
Given the rarity of c.1652G>A in other populations but being the common allele among Thai patients though they were not related and were from different region of Thailand, the best plausible explanation is a founder allele. Nonetheless, we did not find evidence supporting recent common founders up to 40 generations as tested by ERSA. Why Thai and Saskatchewan affected SD patients share the intriguing allele, c.1652G>A, is difficult to answer. Saskatchewan population comprised of several native tribes and later mixed with some French descendants. Given the c.1652G>A variant does not appear to be common among Saskatchewan people, it is possible that recent migration is responsible for the presence of this allele in the population.
HEXB gene is the only gene known to cause SD with approximately 107 variants described with the vast majority being missense and nonsense variants [29]. Most of the HEXB pathologic variants are family-specific but common HEXB variant have been observed in certain ethnic groups such as c.850C>T (p.Arg284*) in Indian population [24], c.171delG (p.Trp57CysfsX6) in Spanish descendants [30], and c.115delG (p.Val39fs) in northern Saskatchewan population of Canada [27]. Possible genotype-phenotype correlation has been suggested, for example c.626C>T (p.Thr209Ile) and c.1404delT (p.P468PfsX62) are likely linked to the infantile form of SD [31]. Our data suggest that the p.Cys551Tyr is associated with severe infantile SD phenotypes.
Our data suggests that SD likely represents the most common subtype of infantile GM2 gangliosidosis identified among Thai patients and that TSD is much less common [7, 8]. Data on incidence/prevalence and carrier frequency of SD and TSD in Asian populations is limited, with only six Chinese [28, 31] and four Japanese cases [32] with infantile onset SD described. The carrier frequency of SD has been well demonstrated in some populations, such as 1 in 310 in Australian [33], 1 in 276 in non-Jewish American [34], and 1 in 15–27 in Saskatchewan [27]. Based on the total of 8,751,131 live births in Thailand during the study period (http://bps.moph.go.th/new_bps/healthdata), the prevalence of infantile SD is estimated at least at 1 in 1,458,521 and carrier frequency at 1 in 604 as ascertained by using Hardy-Weinberg equation. We are convinced that these numbers are far underestimated due to the lack of awareness among physicians and concomitant misdiagnosis.
The identification of the HEXB variants can benefit genetic counseling and prevention of reoccurrence in the affected families, as evidenced by prenatal diagnosis for the subsequent pregnancy of family-2 in the present study. Expanded carrier screening using next generation sequencing for variant detection of serious recessive disorders has been rapidly adopted by couples without existing family history of genetic conditions [35]. Having reference genome of unaffected population and disease-specific variant database of the comparable ethnic group are necessary to increase accuracy of clinically relevant interpretation of the variants identified.
Conclusion
SD is the most common infantile onset-GM2 gangliosidosis among Thai affected population. Biochemical analysis yielded high sensitivity and at low cost, therefore it should be considered as the first diagnostic test for SD. The p.Cys551Tyr is the most common HEXB pathologic allele identified among Thai patients. This data is useful for designing stepwise molecular analysis and for the use of expanded carrier testing.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 4MUG:
-
4-methylumbelliferyl-2-acetomido-2-deoxy-β-D-glucopyranoside
- GM2AP:
-
GM2 activator protein deficiency
- HEX:
-
β-hexosaminidase enzyme
- HEX-A:
-
β-hexosaminidase A
- HEX-B:
-
β-hexosaminidase B
- IBD:
-
identity-by-descent
- IBS:
-
identity-by-state
- SD:
-
Sandhoff disease
- TSD:
-
Tay-Sachs disease
References
Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455(2–3):105–38.
Beutler E, Kuhl W, Comings D. Hexosaminidase isozyme in type O Gm2 gangliosidosis (Sandhoff-Jatzkewitz disease). Am J Hum Genet. 1975;27(5):628–38.
Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54(2):176–82.
Bley AE, Giannikopoulos OA, Hayden D, Kubilus K, Tifft CJ, Eichler FS. Natural history of infantile G(M2) gangliosidosis. Pediatr. 2011;128(5):e1233–41.
Maegawa GH, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, Giugliani R, Mahuran D, Clarke JT. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatr. 2006;118(5):e1550–62.
Sakpichaisakul K, Taeranawich P, Nitiapinyasakul A, Sirisopikun T. Identification of Sandhoff disease in a Thai family: clinical and biochemical characterization. J Med Assoc Thail. 2010;93(9):1088–92.
Wasant P, Wattanaweeradej S, Raksadawan N, Kolodny EH. Lysosomal storage disorders in Thailand: the Siriraj experience. Southeast Asian J Trop Med Public Health. 1995;26(Suppl 1):54–8.
Boonyawat B, Phetthong T, Nabangchang C, Suwanpakdee P. A novel frameshift mutation of HEXA gene in the first family with classical infantile Tay-Sachs disease in Thailand. Neurol Asia. 2016;21:281–5.
Shapira BM, Miller J, Africk D. Hexosaminidase a and B activity (plasma, serum). In: Shapira BM, Miller J, Africk D, editors. Biochemical genetics a laboratory manual. New York Oxford: Oxford University Press; 1989. p. 30–1.
Mukherjee S, Zhang Y. Protein-protein complex structure predictions by multimeric threading and template recombination. Structure. 2011;19(7):955–66.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303.
Maier T, Strater N, Schuette CG, Klingenstein R, Sandhoff K, Saenger W. The X-ray crystal structure of human beta-hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 2003;328(3):669–81.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Loh PR, Danecek P, Palamara PF, Fuchsberger C, Y AR, H KF, Schoenherr S, Forer L, McCarthy S, Abecasis GR et al. Reference-based phasing using the haplotype reference consortium panel. Nat Genet 2016;48(11):1443–1448.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–83.
Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–7.
Gusev A, Lowe JK, Stoffel M, Daly MJ, Altshuler D, Breslow JL, Friedman JM, Pe'er I. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19(2):318–26.
Li H, Glusman G, Hu H, Shankaracharya, Caballero J, Hubley R, Witherspoon D, Guthery SL, Mauldin DE, Jorde LB, et al. Relationship estimation from whole-genome sequence data. PLoS Genet. 2014;10(1):e1004144.
Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343–50.
Abdelhameed TA, Gasmelseed MM, Mustafa MI, Abdelrahman DN, Abdelrhman FA, Hassan MA. Comprehensive Analysis of HEXB Protein Reveal Forty Two Novel nsSNPs That May Lead to Sandhoff disease (SD) Using Bioinformatics. bioRxiv. 2019;853077.
Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep. 2017;7(1):10480.
Tamhankar PM, Mistri M, Kondurkar P, Sanghavi D, Sheth J. Clinical, biochemical and mutation profile in Indian patients with Sandhoff disease. J Hum Genet. 2016;61(2):163–6.
Lee HF, Chi CS, Tsai CR. Early cardiac involvement in an infantile Sandhoff disease case with novel mutations. Brain and Development. 2017;39(2):171–6.
Venugopalan P, Joshi SN. Cardiac involvement in infantile Sandhoff disease. J Paediatr Child Health. 2002;38(1):98–100.
Fitterer B, Hall P, Antonishyn N, Desikan R, Gelb M, Lehotay D. Incidence and carrier frequency of Sandhoff disease in Saskatchewan determined using a novel substrate with detection by tandem mass spectrometry and molecular genetic analysis. Mol Genet Metab. 2014;111(3):382–9.
Wu R, Tang W, Qiu K, Li Y, Lu L, Li D. Analysis of HEXB gene mutations in an infant with Sandhoff disease. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019;36(9):930–4.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136(6):665–77.
Gort L, de Olano N, Macias-Vidal J, Coll MA, Spanish GMWG. GM2 gangliosidoses in Spain: analysis of the HEXA and HEXB genes in 34 Tay-Sachs and 14 Sandhoff patients. Gene. 2012;506(1):25–30.
Zhang W, Zeng H, Huang Y, Xie T, Zheng J, Zhao X, Sheng H, Liu H, Liu L. Clinical,biochemical and molecular analysis of five Chinese patients with Sandhoff disease. Metab Brain Dis. 2016;31(4):861–7.
Fujimaru M, Tanaka A, Choeh K, Wakamatsu N, Sakuraba H, Isshiki G. Two mutations remote from an exon/intron junction in the beta-hexosaminidase beta-subunit gene affect 3′-splice site selection and cause Sandhoff disease. Hum Genet. 1998;103(4):462–9.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. Jama. 1999;281(3):249–54.
Cantor RM, Roy C, Lim JS, Kaback MM. Sandhoff disease heterozygote detection: a component of population screening for Tay-Sachs disease carriers. II. Sandhoff disease gene frequencies in American Jewish and non-Jewish populations. Am J Hum Genet. 1987;41(1):16–26.
Antonarakis SE. Carrier screening for recessive disorders. Nat Rev Genet. 2019;20(9):549–61.
Acknowledgements
We are grateful of the patients’ families and physicians for their participation in this study, Prof. Kanya Suphapeetiporn from Chulalongkorn University for Thai allele frequency data and National Taiwan University Hospital for leukocyte enzyme analysis for a patient. DW, TT and NJ are recipients of the Research Career Development Awards from the Faculty of Medicine Ramathibodi Hospital.
Funding
This study was funded by Mahidol University (to DW). The funding agency played no roles in creating/designing the study and the preparation/writing of the manuscript.
Author information
Authors and Affiliations
Contributions
TT collected and analyzed clinical data, supervised biochemical experiment, and prepared manuscript draft. KW, KK, PT, NV, and KS provided clinical data. AK performed enzyme activity and molecular analysis. ST and TL performed molecular modelling and partly draft the manuscript. KM, NJ, and JE performed PCR-sequencing and/or SNP analysis of the HEXB gene. NS and SM performed array testing. BS performed statistical genetics analysis. DW designed overall concept of the study, supervised data analysis, and critical revision of the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by Ramathibodi Hospital Institutional Review Board (protocol ID 10–51-28) and was conducted in accordance with the ethical standards of national guidelines and with the Helsinki Declaration of 1975, as revised in 2005(5).
Written informed consent was waived because of using archived specimens from service laboratory.
Consent for publication
Yes.
Competing interests
All authors declare that they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplemental Table S1.
Primers sequences and conditions for HEXB mutation analysis. Supplemental Table S2. Scores and prediction of pathogenicity for missense variants identified using in silico analysis tools.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tim-Aroon, T., Wichajarn, K., Katanyuwong, K. et al. Infantile onset Sandhoff disease: clinical manifestation and a novel common mutation in Thai patients. BMC Pediatr 21, 22 (2021). https://doi.org/10.1186/s12887-020-02481-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-020-02481-3