
Abstract
Background
Progressive lung involvement in Filamin A (FLNA)-related cerebral periventricular nodular heterotopia (PVNH) has been reported in a limited number of cases.
Case presentation
We report a new pathogenic FLNA gene variant (c.7391_7403del; p.Val2464Alafs*5) in a male infant who developed progressive lung disease with emphysematous lesions and interstitial involvement. Following lobar resection, chronic respiratory failure ensued necessitating continuous mechanical ventilation and tracheostomy. Cerebral periventricular nodular heterotopia was also present.
Conclusions
We report a novel variant of the FLNA gene, associated with a severe lung disorder and PNVH. The lung disorder led to respiratory failure during infancy and these pulmonary complications may be the first sign of this disorder. Early recognition with thoracic imaging is important to guide genetic testing, neuroimaging and to define optimal timing of potential therapies, such as lung transplant in progressive lung disease.
Similar content being viewed by others

Background
Filamins are large actin-binding proteins that stabilize delicate three-dimensional actin webs and link these to cellular membranes. They integrate cellular architectural and signalling functions and are essential for fetal development and cell locomotion [1].
Filamin A (FLNA) is the first actin filament cross-linking protein identified in non-muscle cells. Mutations in the X-linked gene encoding filamin A (at chromosomal locus Xq28) have been reported to cause a wide range of human diseases, such as cerebral periventricular nodular heterotopia (PVNH), cardiac valvular disease and skeletal anomalies to a variable degree [2,3,4,5,6,7,8,9,10]. Airway anomalies such as tracheal stenosis or tracheobronchomalacia have also been documented and recently lung involvement has been reported [2, 11,12,13,14,15,16,17,18,19,20,21,22].
FLNA-related PVNH is a malformation of cortical development characterized by bilateral near-contiguous ectopic neuronal nodules found along the lateral ventricles [6, 7]. It may be isolated or associated with other brain malformations, including hippocampal malformation and cerebellar hypoplasia, bilateral fronto-perisylvian or temporo-parietooccipital polymicrogyria, hydrocephalus and microcephaly. In a smaller group of patients, PVNH was found to be associated with non-neurologial defects including Ehlers–Danlos syndrome, frontonasal dysplasia, limb abnormalities, ambiguous genitalia and fragile-X syndrome. Finally, several distinct subgroups of patients have been identified with an unusual PVNH presentation, including a micronodular appearance, unilateral distribution and laminar or ribbon-like shapes [23]. Progressive lung involvement in FNLA-related PVNH has been reported in a limited number of cases [2, 11,12,13,14,15,16,17,18,19,20,21,22] and emphysematous lesions in the pulmonary parenchyma are the characteristic findings of this mutation.
We report the case of a male infant with a novel pathogenic variant of the FLNA gene mutation, who developed significant lung disease and in whom a periventricular nodular heterotopia was also diagnosed.
Case presentation
A 32 day old male infant was referred to our department, from another hospital, with acute respiratory distress syndrome and suspected congenital pulmonary malformation. The baby (fourth child of nonconsanguineous caucasian parents) was born by vaginal delivery at 37 weeks’ gestation, with a weight of 3140 g. The first month of life was unremarkable. The family had no history of genetic or metabolic diseases or congenital disorders.
At admission, the physical examination confirmed respiratory distress, general hypotonia due to respiratory failure and fatigue, bilateral inguinal hernia and deformities of the lower limbs (pes tortus congenitalis and hip dysplasia).
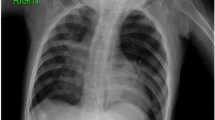
A chest X-ray (Fig. 1) and computed tomography (CT) scan (Fig. 2, Panels a, b) showed severe hyperinflation of the apical segment of the left lung and mediastinal shift to the right. A presumptive diagnosis of congenital lobar emphysema (CLE), including the lower lobe was made. After the stabilization of the subject’s respiratory conditions (non invasive respiratory support, fluid and electrolyte management, broad spectrum antibiotics, bronchodilatator), considering the inclusion of the superior lobe and the upper part of the lower lobe we decided to proceed with observation.

Chest X-ray at admission shows left pulmonary areas of hyperinflation (see arrows)

CT thorax at admission (Panels a, b) and two months later (Panels c, d). The arrows indicate the hyperinflation area. Panels a, c: axial position; Panel b, d: sagittal position
Two months later, the child’s condition deteriorated with worsening in respiratory distress; the child was unable to maintain saturation even with oxygen support. CT-angiography (Fig. 2, Panels c, d) was ordered and revealed a severe lobar emphysema of the anterior to the apicoposterior segment of the left upper lobe, with displacement of mediastinal structures to the right and compression of the right structures. A subsegmental atelectasis and areas of air trapping in the apicoposterior segment of the left lower lobe were also noted. Angiography showed peripheral pulmonary vascular attenuation and central pulmonary artery enlargement.
Surgery included a left upper lobectomy and segmental resection of the left lower lobe. The histopathology report was consistent with a generalized lung growth abnormality with alveolar enlargement and simplification.
Following surgery, multiple attempts to extubate the infant failed and he had a persistent oxygen requirement. Chronic respiratory failure ensued with progressive worsening of the ventilatory performance, necessitating continuous mechanical ventilation, with gradual support parameter adjustments and tracheostomy at age 12 months.
After prolonged multidisciplinary discussion, the decision to perform a surgical thoracoscopic lung biopsy was made in order to obtain additional data on the pathological pulmonary features for prognostic predictions and therapeutic decisions. Histopathology revealed alveolar enlargement, perivascular and interstitial fibrosis and intra-alveolar hemorrhages (Fig. 3).

Histological features. In Panel a, areas in blue and the arrows indicate the perivascular and interstitial fibrosis and intra-alveolar hemorrhages (Azan-Mallory coloration, magnification 10x). In Panel b, areas in brown (Tenascin, magnification 10x) indicate where Tenascin was overexpressed, highlighting the extensive parenchymal fibrosis. TNC localization in the normal lung was un-detectable; TNC is specifically and transiently expressed upon tissue injury and down-regulated when tissue repair or scarring is concluded [38]
Genetic testing was performed during the course of clinical care, after obtaining informed consent. Next generation sequencing on genomic DNA was performed using the NimbleGen SeqCap Target Enrichment kit (Roche) designed to capture several genes involved in pulmonary surfactant protein deficiency and skeletal abnormalities. A library was prepared following the manufacturer’s instructions and subsequently sequenced on an Illumina NextSeq550 instrument. Sequence data were carefully analyzed and the presence of all suspected variants were checked in the public databases (dbSNP, 1000 Genomes, and Exome Aggregation Consortium). The identified variants were confirmed by Sanger sequencing, following a standard protocol (BigDye® Terminator v3.1 Cycle Sequencing Kit,Life Technologies). No potentially causative variants were found in genes associated with cystic fibrosis, pulmonary surfactant protein deficiency or mutations in the SETBP1 gene associated with Schinzel–Giedion syndrome (a rare autosomal dominant disorder that results in facial dysmorphism and organ and bone abnormalities).
Sequencing analysis showed a new mosaic frameshift variant, NM_001456.3: c.7391_7403del, p.Val2464Alafs*5 in the FLNA gene that was not present in the maternal blood DNA. This variant has not been previously reported in individuals with FLNA-related disorders, but can be classified as likely pathogenic (Class 4) according to the ACMG guideline and it is expected to cause disease. It is not present in any public databases, dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/, 1000 Genomes Project (http://www.internationalgenome.org/), EVS (http://evs.gs.washington.edu/EVS/), ExAC (http://exac.broadinstitute.org/) and can be considered as a private variant.
The same mutation was identified in DNA from salivary and pulmonary mesenchymal stem cells of the patient [24].
Brain magnetic resonance imaging (MRI) depicted PVNH (Fig. 4), although the patient was not suffering from any neurological symptoms at this stage.

Brain MRI. Appearance of nodules (indicated by arrows) in periventricular grey matter heterotopia (images b, e, d), surrounding the left temporal horn and merging with the hippocampal cortex (image c). Supratentorial signal alterations with T2 and FLAIR hyperintense (images a, indicated by triangles) as in demyelinating lesions
At 14 months follow-up, the patient requires mechanical ventilation and artificial nutrition to maintain his growth. Epilepsy and other neurological manifestations were not recorded.
Discussion and conclusions
Filamin A is an actin-linking protein that regulates cell shape and migration of many cell types, including neuronal, vascular and cutaneous cells [15]. Filamin A is composed of three main functional domains: (1) a tandem N-terminal calponin-homology domain (CHD1 and CHD2), which confers F-actin binding properties; (2) 15 + 8 internally homologous Ig-like repeats separated by a short run with an unique sequence (hinge 1), important for flexibility; and (3) a second short run (hinge 2) followed by the C-terminal repeat 24, which are important for binding to a wide range of proteins and for dimerization [25]. Null mutations in the FLNA gene, lead to defects in neuronal migration, vascular function and connective tissue integrity. In contrast, gain-of-function missense mutations in this same gene produce a spectrum of malformations in multiple organ systems, especially the skeleton [26].
Here, we report the case of a male child in whom a new mosaic loss-of-function variant of the FLNA gene c.7391_7403del; p.Val2464Alafs*5 was found by next generation sequencing, resulting in significant lung disease characterized by emphysematous lesions and perivascular and interstitial fibrosis. The mutant allele frequency of this variant is estimated to be around 36% considering the numbers of sequence reads of the mutant and the wildtype alleles. This 13 bp deletion is predicted to result in a truncated protein that lacks the hinge 2 domain and repeat 24 probably leading to a loss of binding and dimerization ability that is essential for the FLNA function.
This report confirms an association between a FLNA gene mutation and lung disease. PNVH was observed and limb deformities were also present. There are 25 previous case reports in the literature on FLNA-related disorders with the pulmonary phenotype (Table 1) [2, 9, 13,14,15,16,17,18,19,20,21,22]. Lung diseases are associated with documented PNVH in 84% of the reviewed cases. The presence of cardiac co-morbidities, such as patent ductus arteriosis, valvular disease and aortic root dilatation, have also been reported [2,3,4,5,6,7,8,9,10, 13,14,15, 18]. Mutations in the filamin A gene are inherited in an X-linked (Xq28) dominant manner, with perinatal lethality in most males, whereas in female patients the prognosis depends on the severity of the associated cardiovascular abnormalities [20]. Of the previously published cases 21/25 (80%) were female (Table 1). Perinatal lethality occured in six of these reported cases (24%; 5 females and 1 male); in all cases, cardiopathies were also found [2,3,4,5,6,7,8,9,10, 13,14,15, 18]. As reported in Table 1, a large spectrum of FLNA mutations are detected in patients with pulmonary disease, including missense mutations [9, 13, 14, 19], nonsense mutations [2, 20], deletions [13, 15, 16, 21], duplications [13, 14, 18, 21], truncating mutations [17, 21], and frameshift mutations [14].
In these patients, the presentation of respiratory failure occurred at a median age of 1 month (range, birth to 72 months). However, one reported patient developed progressive obstructive lung disease at the age of 38 years [20]. The clinical presentation of lung involvement was variable, ranging from multiple episodes of intercurrent pulmonary infections [13], to progressive severe pulmonary disease [13, 14, 16,17,18, 20]. A variable outcome and management course were reported in the previously reported cases. In a limited number of patients, supportive therapy was successful [13, 16, 17, 19]. Surgical intervention in the form of lobar resection [2, 9, 13], as in our case, or lung transplantation, may be indicated in severe cases where supportative therapies are not successful [14, 20].
The pulmonary growth abnormality associated with FLNA deficiency consists of multilobar overinflation predominantly affecting the upper and lower lobes, with coarse septal thickening and varying lower lobe atelectasis with pruning of the peripheral pulmonary vasculature [27]. The role of FLNA in the development of lung disease is still not well elucidated. Considering that during respiration the lungs are subjected to mechanical forces and because FLNA plays important role in cell mechanosensing and mechanotransduction, abnormal FLNA interactions could affect pulmonary viscoelastic properties and disturb alveolar formation and growth [14, 28]. However, a role in T cell activation, interleukin production [29], inflammatory signaling [30] and interaction with the cystic fibrosis transmembrane conductance regulator [31] has also been proposed. Furthermore, the crucial role of FLNA action in mesenchymal migration, should not be excluded. Alterations in mesenchymal properties could be directly related to defects in cell migration during embryonic development and in pulmonary damage described in FLNA-defective patients [32]. Further studies are needed to investigate the functional role of tissue-resident lung mesenchymal stem cells in health and disease. Considering the successful use of stem cell therapy in the treatment of chronic progressive pulmonary disease in adults [31,32,33,34,35,36,37], future perspective stem cell treatment also in FLNA mutation-related lung disorders in children should be investigated. In conclusion, we report a novel mosaic loss-of-function variant of the FLNA gene associated with a severe lung disorder and PNVH. The lung disorder led to respiratory failure during infancy and these pulmonary complications may be the first sign of this disorder. Early recognition with thoracic imaging is important to guide genetic testing, neuroimaging and to define optimal timing of potential therapies, such as lung transplant in progressive lung disease [14].
Abbreviations
- CLE:
-
Congenital lobar emphysema
- CT:
-
Computed tomography
- FLNA:
-
Filamin A
- PVNH:
-
Periventricular nodular heterotopia
References
Stossel TP. Filamins and the potential of complexity. Cell Cycle. 2010;9:1463.
Masurel-Paulet A, Haan E, Thompson EM, Goizet C, Thauvin-Robinet C, Tai A, Kennedy D, Smith G, Khong TY, Solé G, Guerineau E, Coupry I, Huet F, Robertson S, Faivre L. Lung disease associated with periventricular nodular heterotopia and an FLNA mutation. Eur J Med Genet. 2011;54:25–8.
Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–8.
Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–25.
Roberson S, Twigg S, Sutherland-Smith A, Biancalana V, Gorlin R, Horn D, et al. Localized mutations in the gene encoding the cytoskeletal protein filamin a cause diverse malformations in humans. Nat Genet. 2003;33:487–91.
Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10:1775–83.
Sheen V, Jansen A, Chen M, Parrini E, Morgan T, Ravenscroft R, et al. Filamin a mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–62.
de Wit M, Kros J, Halley D, de Coo I, Verdijk R, Jacobs B, et al. Filamin a mutation, a common cause for periventricular heterotopia, aneurysms and cardiac defects. J Neurol Neurosurg Psychiatry. 2009;80:426–8.
de Wit MC, Tiddens HA, de Coo IF, Mancini GM. Lung disease in FLNA mutation: confirmatory report. Eur J Med Genet. 2011;54:299–300.
Demirel N, Ochoa R, Dishop MK, Holm T, Gershan W, Brottman G. Respiratory distress in a 2-month-old infant: Is the primary cause cardiac, pulmonary or both? Respir Med Case Rep. 2018;25:61-5.
Lord A, Shapiro AJ, Saint-Martin C, Claveau M, Melançon S, Wintermark P. Filamin a mutation may be associated with diffuse lung disease mimicking bronchopulmonary dysplasia in premature newborns. Respir Care. 2014;59:e171–7.
Lu J, Sheen V. Periventricular heterotopia. Epilepsy Behav. 2005;7:143–9.
Shelmerdine SC, Semple T, Wallis C, Aurora P, Moledina S, Ashworth MT, Owens CM. Filamin a (FLNA) mutation-a newcomer to the childhood interstitial lung disease (ChILD) classification. Pediatr Pulmonol. 2017;52:1306–15.
Burrage LC, Guillerman RP, Das S, Singh S, Schady DA, Morris SA, Walkiewicz M, Schecter MG, Heinle JS, Lotze TE, Lalani SR, Mallory GB. Lung transplantation for FLNA-associated progressive lung disease. J Pediatr. 2017;186:118–123.e6.
Kinane TB, Lin AE, Lahoud-Rahme M, Westra SJ, Mark EJ. Case 4-2017. A 2-month-old girl with growth retardation and respiratory failure. N Engl J Med. 2017;376:562–74.
Sassaki E, Byrne AT, Phelan E, Cox DW, Reardon W. A review of filamin a mutations and associated interstitial lung disease. Eur J Pediatr. 2018. https://doi.org/10.1007/s00431-018-3301-0.
Hathorn C, Armitage N, Wensley D, Seear M. Bronchial balloon occlusion in children with complex pulmonary air leaks. Arch Dis Child. 2013;98:136-40.
Eltahir S, Ahmad KS, Al-Balawi MM, Bukhamsien H, Al-Mobaireek K, Alotaibi W, Al-Shamrani A. Lung disease associated with filamin a gene mutation: a case report. J Med Case Rep. 2016;10:97.
Gérard-Blanluet M, Sheen V, Machinis K, Neal J, Apse K, Danan C, Sinico M, Brugières P, Mage K, Ratsimbazafy L, Elbez A, Janaud JC, Amselem S, Walsh C, Encha-Razavi F. Bilateral periventricular heterotopias in an X-linked dominant transmission in a family with two affected males. Am J Med Genet A. 2006;140:1041–6.
Lange M, Kasper B, Bohring A, Rutsch F, Kluger G, Hoffjan S, Spranger S, Behnecke A, Ferbert A, Hahn A, Oehl-Jaschkowitz B, Graul-Neumann L, Diepold K, Schreyer I, Bernhard MK, Mueller F, Siebers-Renelt U, Beleza-Meireles A, Uyanik G, Janssens S, Boltshauser E, Winkler J, Schuierer G, Hehr U. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis. 2015;10:134.
Clapham KR, Yu TW, Ganesh VS, Barry B, Chan Y, Mei D, Parrini E, Funalot B, Dupuis L, Nezarati MM, du Souich C, van Karnebeek C, Guerrini R, Walsh CA. FLNA genomic rearrangements cause periventricular nodular heterotopia. Neurology. 2012;78:269–78.
Reinstein E, Chang BS, Robertson SP, Rimoin DL, Katzir T. Filamin a mutation associated with normal reading skills and dyslexia in a family with periventricular heterotopia. Am J Med Genet A. 2012;158A:1897–901.
Parrini E, Ramazzotti A, Dobyns WB, Mei D, Moro F, Veggiotti P, Marini C, Brilstra EH, Dalla Bernardina B, Goodwin L, Bodell A, Jones MC, Nangeroni M, Palmeri S, Said E, Sander JW, Striano P, Takahashi Y, Van Maldergem L, Leonardi G, Wright M, Walsh CA, Guerrini R. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin a mutations. Brain. 2006;129:1892–906.
Calcaterra V, Avanzini MA, Mantelli M, Agolini E, Croce S, De Silvestri A, Re G, Collura M, Maltese A, Novelli A, Pelizzo G. A case report on filamin A gene mutation and progressive pulmonary disease in an infant: A lung tissued derived mesenchymal stem cell study. Medicine (Baltimore). 2018;97:e13033.
Parrini E, Guerrini R. Lissencephaly type I and periventricular heterotopia. In Encyclopedia of Neuroscience. Elsevier Ltd.; 2010. p. 503–12. https://doi.org/10.1016/B978-008045046-9.01503-5.
Robertson SP. Filamin a: phenotypic diversity. Curr Opin Genet Dev. 2005 Jun;15(3):301–7.
Semple TR, Ashworth MT, Owens CM. Interstitial lung disease in children made easier…well. Almost Radiographics. 2017;37:1679–703.
Razinia Z, Mäkelä T, Ylänne J, Calderwood DA. Filamins in mechanosensing and signaling. Annu Rev Biophys. 2012;41:227–46.
Hayashi K, Altman A. Filamin a is required for T cell activation mediated by protein kinase C-θ. J Immunol. 2006;177:1721–8.
Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U. Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2000;275:271–8.
Thelin WR, Chen Y, Gentzsch M, Kreda SM, Sallee JL, Scarlett CO, et al. Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR. J Clin Invest. 2007;117:364–74.
Guiet R, Vérollet C, Lamsoul I, Cougoule C, Poincloux R, Labrousse A, Calderwood DA, Glogauer M, Lutz PG, Maridonneau-Parini I. Macrophage mesenchymal migration requires podosome stabilization by filamin a. J Biol Chem. 2012;287:13051–62.
Lian G, Kanaujia S, Wong T, Sheen V. FilaminA and Formin2 regulate skeletal, muscular, and intestinal formation through mesenchymal progenitor proliferation. PLoS One. 2017;12:e0189285.
Mohammadipoor A, Antebi B, Batchinsky AI, Cancio LC. Therapeutic potential of products derived from mesenchymal stem/stromal cells in pulmonary disease. Respir Res. 2018;19:218.
Kokturk N, Yıldırım F, Gülhan PY, Oh YM. Stem cell therapy in chronic obstructive pulmonary disease. How far is it to the clinic? Am J Stem Cells. 2018;7:56–71.
Broekman W, Khedoe PPSJ, Schepers K, Roelofs H, Stolk J, Hiemstra PS. Mesenchymal stromal cells: a novel therapy for the treatment of chronic obstructive pulmonary disease? Thorax. 2018;73:565–74.
Sun Z, Li F, Zhou X, Chung KF, Wang W, Wang J. Stem cell therapies for chronic obstructive pulmonary disease: current status of pre-clinical studies and clinical trials. J Thorac Dis. 2018 Feb;10(2):1084–98.
Estany S, Vicens-Zygmunt V, Llatjós R, Montes A, Penín R, Escobar I, Xaubet A, Santos S, Manresa F, Dorca J, Molina-Molina M. Lung fibrotic tenascin-C upregulation is associated with other extracellular matrix proteins and induced by TGFβ1. BMC Pulm Med. 2014;14:120.
Acknowledgments
The authors thank Dr. L. Kelly for English revision of the manuscript.
Funding
The authors declare that they did not receive any source of funding for the preparation of the manuscript.
Availability of data and materials
This section is not applicable.
Author information
Authors and Affiliations
Contributions
GP, GC management of the patient, drafting the article, critical revision of the article; MC, AP, MPP, CC management of the patient, critical revision of the article; EA, AN, MP genetic evaluation, drafting the article, critical revision of the article; RB histological evaluation; VC drafting the article, literature review, critical revision of the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
The study was performed according to the Declaration of Helsinki. Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Competing interests
The authors have no competing interests to declare.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pelizzo, G., Collura, M., Puglisi, A. et al. Congenital emphysematous lung disease associated with a novel Filamin A mutation. Case report and literature review. BMC Pediatr 19, 86 (2019). https://doi.org/10.1186/s12887-019-1460-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1460-4