
Abstract
Background
Well-differentiated (WD) neuroendocrine tumors (NETs) are a group of rare neoplasms with limited therapeutic options. Cabozantinib is an inhibitor of multiple tyrosine kinases with a pivotal role in NET pathogenesis, including c-MET and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2). LOLA is the first prospective phase II trial aiming to assess the safety and activity of cabozantinib combined with lanreotide in WD NETs of gastroenteropancreatic (GEP), thoracic and of unknown origin.
Methods
This is a multicenter, open-label, double-cohort, non comparative, non-randomized, three-stage phase II trial. Eligible patients have to meet the following inclusion criteria: diagnosis of advanced or metastatic, progressive, non-functioning WD thoracic NETs, GEP-NETs or NETs of unknown origin with Ki67 ≥ 10%; positive 68 Ga-PET uptake or somatostatin receptor 2 immunohistochemical (IHC) stain; maximum 1 prior systemic regimen for metastatic disease. Two cohorts will be considered: pNETs and carcinoids (typical or atypical lung and thymus NETs, gastro-intestinal NETs or NETs of unknown origin). In stage I, the primary objective is to find the optimal dose of cabozantinib in combination with lanreotide and to evaluate the safety of the combination (percentage of patients experiencing grade 3–5 toxicities according to NCI-CTCAE version 5.0). Starting dose of cabozantinib is 60 mg/day continuously, plus lanreotide 120 mg every 28 days. In stage II and III, co-primary endpoints are safety and overall response rate (ORR) according to RECIST version 1.1. The uninteresting antitumor activity is fixed in ORR ≤ 5%. Secondary endpoints are progression-free survival and overall survival. Exploratory objectives include the assessment of c-MET, AXL and VEGFR2 IHC expression, to identify predictive or prognostic tissue biomarkers. Enrolment started in July 2020, with an expected trial duration of 42 months comprehensive of accrual, treatment and follow-up. Considering a drop-out rate of 5%, the maximum number of enrolled patients will be 69.
Discussion
Supported by a solid rationale, the trial has the potential to generate milestone data about the synergistic effects of cabozantinib plus lanreotide in a group of NET patients with relatively aggressive disease and limited therapeutic options.
Trial registration
LOLA is registered at ClinicalTrials.gov (NCT04427787) and EudraCT (2019–004506-10).
Similar content being viewed by others

Background
Neuroendocrine tumors (NETs) are rare malignant neoplasms that arise from diffuse neuroendocrine cells. As a result of increased detection rate, improved diagnostic accuracy and advances in systemic therapies, the annual age-adjusted incidence of NETs increased from 1.09/100000 persons per year in 1973 to 6.98/100000 in 2012, with a parallel rise in prevalence [1].
Approximately 80 to 90% of well-differentiated (WD) gastroenteropancreatic (GEP) and thoracic NETs express somatostatin receptors (SSTRs) on their cell surface, that bind with high affinity somatostatin analogs (SSAs) lanreotide autogel and octreotide long-acting release (LAR). These drugs are the mainstay for the control of NET-associated endocrine syndromes and represent one pivotal standard treatment option for patients with advanced NETs expressing SSTRs [2,3,4]. Based on primary site and histopathological features, WD NETs can display heterogeneous clinical behavior and prognosis, grade (G)2 and G3 NETs being more aggressive than the indolent G1 forms [5]. New combinations of SSAs and other investigational drugs are therefore warranted, with the aim to improve clinical outcomes, while maintaining a good tolerability profile.
Combined inhibition of receptor tyrosine kinases and angiogenesis has already proven an effective strategy in the treatment of advanced NETs. The multikinase inhibitors sunitinib and pazopanib—targeting Vascular Endothelial Growth Factor Receptor (VEGFR) 2–3, platelet-derived growth factor receptors (PDGFR α/β), and c-KIT—showed clinical activity in advanced NETs. Sunitinib has been approved for the treatment of advanced pancreatic (p)NETs based on the results of a phase III randomized clinical trial [6,7,8].
Current research is increasingly focusing on the development of new multitarget tyrosine kinase inhibitors (TKIs), with the aim to improve therapeutic efficacy. Surufatinib (a VEGFR 1–3, fibroblast growth factor receptor (FGFR) -1 and colony-stimulating-factor-1 receptor inhibitor) has recently shown a progression-free survival (PFS) advantage over placebo in two randomized phase III trials in advanced NETs of pancreatic and extra-pancreatic origin [9, 10]. Similarly lenvatinib (a VEGFR 1–3, FGFR1-4, PDGFRα, RET and KIT inhibitor) showed interesting activity in pre-treated WD NET patients in a phase II trial, whereas axitinib (a VEGFR 1–3 inhibitor) combined with octreotide LAR improved response rates and PFS outcomes over placebo in a randomized phase II/III study [11, 12]. More recently, results of the prospective randomized placebo- controlled phase II Alliance A021202 trial of pazopanib (an oral VEGFR-2,-3, PDGFR-α, and β, and c-KIT inhibitor) in patients with progressive carcinoid tumors demonstrated a PFS advantage in the interventional arm over the placebo arm [13].
Cabozantinib is an orally administered small molecule that inhibits the enzymatic activity of multiple tyrosine kinases including c-MET, VEGFR2, RET, KIT, AXL, and Fms Related Tyrosine Kinase 3 [14]. Recent studies suggest that c-MET signaling may play a role in NET biology and pathogenesis. c-MET protein overexpression and increased mRNA levels were detected in 17–33% of primary GEP-NETs and in up to 60% of metastatic sites. c-MET levels correlate with increased Ki-67 proliferation index, increased risk of developing liver metastases and decreased survival in GEP NETs [15,16,17,18]. c-MET and/or its phosphorylated form were also described in typical and atypical lung carcinoids, with more than 80% of cases showing a strong expression [19].
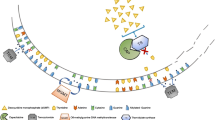
The biological rationale of the synergistic effects of SSAs in combination with cabozantinib resides in the concomitant inhibition of intracellular signaling pathways associated with tumor cell proliferation, angiogenesis and immune modulation. The antiproliferative effects of SSAs are mediated by the inhibition of the Ras/mitogen-activated protein kinase (MAPK) and phosphoinositide 3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) cascades. In vitro, SSTR2 enhances the dephosphorylation of PI3K and Akt, negatively affecting mTOR-mediated protein synthesis and translation. Moreover, the simultaneous stimulation of SSTR 2 and 5 achieves significant inhibition of Ras and extracellular signal-regulated kinase (ERK)-1/2. [20,21,22,23] Similarly, cabozantinib inhibits the activity of VEGFR2, c-MET and AXL at the level of both cancer cells and peritumoral vessels, thus leading to the downstream inhibition of PI3K/Akt/mTOR and Ras/MAPK, with antiproliferative and antiangiogenic effects. [24] Also SSAs exert anti-angiogenic effects by engaging SSTRs on the vascular endothelium and by downregulating the secretion of proangiogenic factors, such as VEGF and PDGF [25]. Immune modulation has been advocated as a part of the antitumor effect of several targeted therapies, including cabozantinib and SSAs. Cabozantinib treatment in combination with SSAs may therefore modulate circulating immune populations and the tumor immune infiltrate, potentially reverting the tumor-associated immunosuppressive phenotype, and strengthening the biological rationale of exploiting this combination in NETs [26,27,28,29,30,31] (Fig. 1).

Synergistic antiproliferative, anti-angiogenetic and immune-modulatory effects of cabozantinib combined with somatostatin analogs. cAMP: cyclic adenosine monophosphate; ERK: extracellular signal-regulated kinases; HGF: hepatocyte growth factor; NFkB: nuclear factor kappa-light-chain-enhancer of activated B cells; MDSC: myeloid-derived suppressor cell; mTOR: mammalian target of rapamycin; PI3K: phosphoinositide 3 kinase; RAS: Rat sarcoma virus; RAF: Rapidly Accelerated Fibrosarcoma; SHP1: Src homology region 2 domain-containing phosphatase-1; SSA: somatostatin analog; SSTR: somatostatin receptor; TCL: T cell Lymphocyte; Treg: regulatory T cell; VEGF(R): Vascular Endothelial Growth Factor (Receptor)
The safety and the activity of cabozantinib in NETs has been assessed in a recent phase II trial, that evaluated cabozantinib at the dose of 60 mg/day in 61 patients with progressive, WD G1-2 carcinoids (41 patients) and pNETs (20 patients). Overall response rate (ORR), which represented the primary endpoint of the study, was 15% both in the pNET (95% Confidence Interval [CI] 5–36%) and carcinoid groups (ORR 15%, 95% CI: 7–28%). Median (m) PFS was 21.8 months (95% CI: 8.5–32.0) in patients with pNETs and 31.4 months (95% CI: 8.5- not reached) in patients with carcinoids. Although dose reductions were required in 81% of the 53 patients completing more than 1 treatment cycle, treatment proved tolerable. The most frequent G 3/4 toxicities included hypertension (13%), hypophosphatemia (11%) and diarrhea (10%) [14].
Based on these encouraging preliminary data, further evaluation of cabozantinib in advanced NET patients is warranted. Exploiting new combinations of SSAs and targeted agents, such as cabozantinib, with potential synergistic activity represents an issue of particular clinical interest especially in the relatively high-risk population of G2-G3 WD NET and thoracic carcinoids, for whom therapeutic options are limited and SSA monotherapy may be insufficient to guarantee adequate and durable disease control.
The LOLA trial is the first prospective phase II study aiming to assess the safety and activity of cabozantinib in combination with lanreotide in WD G2-G3 NETs of GEP and of unknown origin with Ki-67 ≥ 10% and in thoracic carcinoids, with the aim to evaluate as secondary endpoints the impact of this combination on PFS and overall survival (OS).
Here we report the methodological study protocol of the LOLA trial (NCT04427787).
Methods
Study design
This is a multicenter, open-label, double cohort, non comparative, non-randomized, three-stage phase II trial aiming to assess the safety and activity of the combination of cabozantinib and lanreotide in patients with unresectable, advanced or metastatic WD G2-3 GEP-NETs with Ki-67 ≥ 10% (including pancreatic NET, small intestine NET, stomach NET, colon and rectum NET), thoracic NETs (including typical and atypical carcinoids of the lung and thymus) independently of proliferation index and NETs of unknown origin with Ki-67 ≥ 10%.
A list of participating Centers is reported in Table 1.
Study population
Adult patients (male or female) that are 18 years of age or older will be enrolled according to the following inclusion criteria: diagnosis of unresectable, advanced or metastatic non-functioning WD GEP-NET (including G2-G3 pNET, small intestine NET, stomach NET, colon and rectum NET) with Ki-67 ≥ 10%, WD thoracic NET (typical and atypical carcinoids of the lung and thymus) independently of proliferation index, or WD NET of unknown primary with Ki-67 ≥ 10%; locally advanced or metastatic disease documented as progressive by response evaluation criteria in solid tumors (RECIST) version 1.1. on computed-tomography (CT)-scan or magnetic resonance imaging (MRI) within 12 months prior to baseline [32]; documented Octreoscan/positron emission tomography (PET) Ga68 uptake according to the Krenning score [33] within 6 months before study entry or SSTR2 immunohistochemical stain [34] on pretreatment/archival tumor tissue samples; Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 or 1; treatment naïve patients or patients who have received maximum 1 prior systemic regimen for metastatic disease (prior peptide receptor radionuclide therapy must be completed at least 6 months prior to enrolment; prior treatment with SSAs, biologic therapy including everolimus or TKIs, immunotherapy, chemotherapy, ablative therapies and/or radiation must be completed at least 28 days prior to registration); adequate liver, kidney, bone marrow and thyroid function.
Patients must not meet any of the following exclusion criteria: poorly differentiated neuroendocrine carcinomas; prior treatment with dose superior or equal to 120 mg per month of lanreotide; prior treatment with cabozantinib; known brain metastases or cranial epidural disease unless adequately treated with radiotherapy and/or surgery, stable for at least 3 months before study entry and neurologically asymptomatic in the absence of corticosteroids; history of cardiac angioplasty or stenting, myocardial infarction, unstable angina, symptomatic peripheral vascular disease, New York Heart Association Class III or IV congestive heart failure, cerebrovascular accidents, active bleeding or bleeding diathesis, pulmonary embolism or untreated deep venous thrombosis within the past 6 months; history of aneurysms and arterial dissections; poorly controlled hypertension.
Study treatment
Cabozantinib will be administered orally at the starting dose of 60 mg/day continuously, in combination with lanreotide 120 mg administered through deep subcutaneous injection every 28 days. Both treatments will start the same day. When cabozantinib dose reduction is necessary, it is recommended to reduce from 60 mg daily to 40 mg daily, and then to 20 mg daily. The dose of lanreotide will be fix thorough the study (standard dose).
Study objectives and endpoints
The primary objective of the study is to demonstrate the safety, tolerability and activity of the association of cabozantinib and lanreotide in patients with WD GEP-NETs, thoracic NETs and NETs of unknown primary. Trial design consists of three stages.
In the I (run-in) stage, the primary objective is to evaluate the safety and tolerability of the combination of cabozantinib plus lanreotide, defined as the percentage of patients experiencing G3-5 toxicities according to National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0.
In II and III stage co-primary objectives will be: a. activity of the combination of cabozantinib plus lanreotide in terms of ORR, defined as the proportion of patients whose confirmed best overall response is either a partial response or complete response, based on investigator-assessed RECIST criteria version 1.1; b. safety and tolerability of the combination defined as percentage of G3-5 toxicities according to NCI-CTCAE version 5.0.
Secondary objectives (stages II and III) will be to evaluate clinical efficacy of cabozantinib in combination with lanreotide in terms of: a. PFS, defined as the time from first day of treatment administration until disease progression according to RECIST criteria version 1.1 or death from any cause, whichever occurs first; b. OS, defined as the time from first day of treatment administration until death from any cause.
Translational objectives
Exploratory objectives include the investigation of the prognostic and predictive role of tissue biomarkers. As mandatory inclusion criteria, representative formalin fixed paraffin embedded tissue (either archival or recently biopsied prior to study entry) collected from the primary tumor and/or a metastatic site will be analyzed by immunohistochemistry to determine VEGFR2, MET, AXL expression levels and their association with ORR and time-to-event endpoints (i.e. PFS and OS). In addition, tumor tissue may be used to explore other possible predictors of outcomes in WD NETs receiving lanreotide plus cabozantinib, with the aim to select and identify tissue biomarkers modulating treatment activity and/or safety.
Sample size and statistical design
The statistical design is adaptive, according to three sequential stages (see the study flow-chart depicted in Fig. 2).

Study design N: number; pNETs: pancreatic neuroendocrine tumors; ORR: Overall response rate; OS: overall survival; PFS: progression-free survival; WD: well-differentiated
Two cohorts of patients will be enrolled in the study according to different histotypes: the first cohort includes pNETs; the second cohort includes carcinoids (i.e. small intestine NETs, stomach NETs, colon and rectum NETs, typical and atypical lung and thymus NETs) and NETs of unknown origin. Enrolment in the pancreatic and carcinoid arms will be competitive in the first stage (i.e. run-in stage).
I stage. The primary objective of I stage is to find the optimal dose of cabozantinib in combination with lanreotide and to evaluate the safety and the tolerability of the combination. Starting dose of cabozantinib is 60 mg daily plus lanreotide 120 mg every 28 days. No more than 7 patients will be enrolled in the I stage. If G3-5 adverse events (AEs)—excluded not clinically relevant laboratory abnormalities according to NCI-CTCAE version 5.0 or clinically manageable and reversible G3 AEs within 7 days of onset—are observed in more than one patient out of the first seven enrolled patients, cabozantinib starting dose of 60 mg will be considered too toxic and ruled out (H0: % grade 3–5 toxicities ≤ 5%; nominal p-value: 0.05; actual p-value: 0.044) and 40 mg will be chosen as starting dose of cabozantinib in the subsequent stage. Otherwise, 60 mg will be chosen as starting dose of cabozantinib in the subsequent stage. An interim safety analysis will be performed when 7 patients will be observed on treatment for at least 8 weeks.
In order to preserve internal validity, outcomes’ assessment will be re-started de novo in the II stage (i.e. I and II stages are distinct phases of an operationally seamless, not inferentially seamless design).
II-III stage. In stages II-III the optimal Simon two-stage design will be used for both cohorts [35]. The primary endpoint of the II and III stages is ORR according to RECIST version 1.1. Co-primary endpoint will be the evaluation of the percentage of G 3–5 toxicities according to NCI-CTCAE version 5.0. Secondary endpoints will be PFS and OS.
The uninteresting antitumor activity (H0) is fixed in ORR ≤ 5%. The useful antitumor activity to be detected (H1) is fixed in ORR ≥ 20%. For both arms (pNET and carcinoids), 10 patients will be accrued in the stage II. If no responses are observed in these 10 patients, the arm will be stopped (nominal type II error = 0.20). Otherwise, other 19 patients will be accrued in stage III, for a total of 29 patients. The null hypothesis will be rejected if 4 or more responses are observed in 29 eligible patients (nominal type I error = 0.05). Considering a drop-out rate of 5% (i.e. eligible patients without the assessment of objective response) the maximum number of enrolled patients will be 69 (including the 7 patients included in the interim analysis after stage I).
Regarding the safety co-primary endpoint, the null hypothesis to be rejected is a percentage of AEs greater or equal to 20% (i.e. H0: ≥ 20%, nominal type I error = 0.05) against an alternative hypothesis H1: ≤ 5%. Because the statistical design is adaptive (i.e. a couple of Simon’s two stage designs are planned after the first run-in stage), the total number of patients that could be analyzed for safety is uncertain prior to study execution. It will be 20 patients if both arms stop at stage II, 39 patients if only one arm stops at stage II and 58 patients if both arms participate to stage III. Thresholds to identify significant level at 5% and exact binomial test’s power will be determined based on the real number of patients to be analyzed (i.e. 0 out of 20 patients [test’s power: 36%], 3 out of 39 patients [test’s power: 87%]; 6 out of 58 patients [test’s power: 97%] are the maximum number of patients with observed G3-G5 AEs to reject H0: ≥ 20% at a 5% significant level). Patients who did not receive treatment for any cause or severely violated protocol inclusion/exclusion criteria will be excluded from the primary and secondary analyses of all stages.
Study procedures
Baseline assessments, including a 12-lead electrocardiogram (ECG) with QTc interval evaluation, a multigated acquisition (MUGA) scan or cardiac echocardiogram and radiological evaluation (CT or MRI of the chest, abdomen and pelvis) should be obtained within 28 days prior to start of study treatment. All additional suspected sites of disease (brain, bone) should be imaged at baseline.
Disease assessment will be performed every 3 cycles (time window for the scans is ± 28 days) with the same imaging modalities used at baseline. A blinded Central Review of imaging will be performed.
Subjects will be monitored continuously for AEs while on study from the time of signing informed consent through 30 days after the date of the decision to permanently discontinue study treatment.
All patients who are discontinued from study drug for any reason other than disease progression will continue to have tumor assessments as per study schedule until documented disease progression or until the initiation of new anticancer therapy. Table 2 reports study procedures and assessments.
Safety
All AEs that occur between the first study-related procedure and 30 days after the last dose of study drug will be reported in the source document and in electronic Case Report Forms. Serious adverse events (SAEs) are defined as any AE that results in death, that is life threatening, that places the subject at immediate risk of death or that results in hospitalization, prolongation of existing hospitalization or in a persistent significant disability. All SAEs that occur following the subject’s written consent to participate in the study through 90 days of discontinuation of dosing must be reported to the Sponsor within 24 h after learning of the event, regardless of relationship to study treatment. Expedited reporting will follow local and international regulations.
Safety data will be reviewed by Data Monitoring Committee on a periodic basis, approximately every 3 months from the date of first-patient-in.
Ethics and regulatory considerations
The study is conducted in full conformance with the International Conference on Harmonisation (ICH) E6 guideline for Good Clinical Practice (GCP), the principles of the Declaration of Helsinki and according to the laws and regulations of the country in which the research is conducted.
The study has been approved by the Institutional Review Board/Independent Ethics Committee (IRB/IEC) of the Coordinating Center (Fondazione IRCCS Istituto Nazionale dei Tumori di Milano) and by the local Ethics Committees of participating centers. The study has been registered in EudraCT database (2019–004506-10) and at clinicaltrials.gov (NCT04427787). Written informed consent will be obtained as a prerequisite for study entry.
Recruitment
Enrolment started in July 2020 and is currently ongoing, with an expected trial duration of 42 months comprehensive of accrual, treatment and follow-up.
Discussion
Cabozantinib is an oral multitarget TKI approved for the treatment of advanced hepatocellular carcinoma (HCC) in patients who have previously been treated with sorafenib, for advanced or metastatic differentiated thyroid carcinoma in patients refractory or not eligible to radioactive iodine who have progressed on prior systemic therapy, and for advanced intermediate or poor risk renal cell carcinoma (RCC)—as first-line treatment or following prior VEGF-targeted therapy [36,37,38,39]. In the advanced RCC setting, cabozantinib was shown to improve PFS outcomes compared to everolimus and sunitinib in the pretreated and first-line settings, respectively [36, 37].
Sunitinib is the only multitargeted TKI approved for advanced pNETs [6]. Similarly to sunitinib, cabozantinib inhibits the tyrosine kinase enzymatic activities of multiple receptors including VEGFR2, MET, AXL, and RET, which have been implicated in tumor proliferation, survival and neoangiogenesis [14, 24, 25]. Based on the efficacy and activity data from phase II and III trials in RCC, HCC, thyroid carcinoma and NET patients [14, 36,37,38,39], due to its high-spectrum biological activity against multiple and non-redundant oncogenic pathways, cabozantinib may be superior to other angiogenesis inhibitors and provide clinical benefit in the treatment of rare tumors with few available treatment options, such as NETs. Other multitarget TKIs (surufanitib, lenvatinib, pazopanib) have recently shown promising activity and efficacy in patients with metastatic NETs, thus corroborating the investigation of such agents in this setting [9,10,11, 13]. The activity of single-agent cabozantinib has been investigated in a phase II study in G1-2 carcinoids and pNETs, and this agent is currently being investigated in patients with advanced NETs progressing on prior therapy in the phase III randomized placebo-controlled CABINET trial (NCT03375320). However, to date, no specific data have been published with regard to the relatively high-risk population of G2-G3 WD NET, for whom combinations of SSAs and potentially synergistic targeted agents represent an unmet clinical need.
In our opinion, the combination of cabozantinib with other agents such as SSAs may result in synergistic antiproliferative effects, potentially enhancing antitumor activity and clinical outcomes. The biological rationale of the synergistic effects of cabozantinib in combination with SSAs resides in the concomitant inhibition of intracellular signaling pathways associated with tumor cell proliferation, angiogenesis and immune modulation [15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31].
LOLA is the first multicenter, open-label, phase II trial aiming to prospectively define the safety and activity of a concomitant treatment with cabozantinib and lanreotide in progressive advanced WD thoracic NETs, G2-3 GEP-NETs and NETs of unknown origin with Ki-67 ≥ 10%. Supported by a solid biological and clinical rationale, the trial has the potential to generate milestone data about the possible synergistic effects of this combination in a population of NET patients with relatively aggressive disease, for whom therapeutic options are limited and an optimal treatment sequence is not yet defined. Moreover, biomarkers’ investigation may allow to identify prognostic tools associated with treatment benefit to better inform patient’s selection for this treatment strategy.
Availability of data and materials
Not applicable.
Abbreviations
- AEs:
-
adverse events
- CI:
-
Confidence Interval
- CT:
-
computed-tomography
- ECG:
-
electrocardiogram
- ECOG:
-
Eastern Cooperative Oncology Group
- ERK:
-
extracellular signal-regulated kinase
- FGFR:
-
fibroblast growth factor receptor
- G:
-
grade
- GEP:
-
gastroenteropancreatic
- HCC:
-
hepatocellular carcinoma
- ICH:
-
GCP International Conference on Harmonisation E6 guideline for Good Clinical Practice
- IRB/IEC:
-
Institutional Review Board/Independent Ethics Committee
- m:
-
Median
- MAPK:
-
Mitogen-activated protein kinase
- MRI:
-
Magnetic resonance imaging
- MTC:
-
Medullary thyroid cancer
- mTOR:
-
Mammalian target of rapamycin
- MUGA:
-
Multigated acquisition
- NCI-CTCAE:
-
National Cancer Institute-Common Terminology Criteria for Adverse Events
- NETs:
-
Neuroendocrine tumors
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- p:
-
Pancreatic
- PDGF:
-
Platelet-derived growth factor
- PET:
-
Positron emission tomography
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphoinositide 3 kinase
- PS:
-
Performance status
- RCC:
-
Renal cell carcinoma
- RECIST:
-
Response evaluation criteria in solid tumors
- SAEs:
-
Serious adverse events
- SSAs:
-
Somatostatin analogs
- SSTRs:
-
Somatostatin receptors
- TKIs:
-
Tyrosine kinase inhibitors
- VEGF:
-
Vascular Endothelial Growth Factor Receptor
- WD:
-
Well-differentiated
References
Dasari A, Shen C, Halperin D, et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017;3(10):1335–42.
Pavel M, de Herder WW. ENETS Consensus Guidelines for the Standard of Care in Neuroendocrine Tumors. Neuroendocrinology. 2017;105(3):193–5.
Rinke A, Muller H, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID study group. J Clin Oncol. 2009;27(28):4656–63.
Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(16):1556–7.
Perren A, Couvelard A, Scoazec JY, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Pathology: diagnosis and prognostic stratification. Neuroendocrinology. 2017;105:196–200.
Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–13.
Grande E, Capdevila J, Castellano D, et al. Pazopanib in pretreated advanced neuroendocrine tumors: a phase II, open-label trial of the spanish task force group for neuroendocrine tumors (GETNE). Ann Oncol. 2015;26(9):1987–93.
Phan AT, Halperin DM, Chan JA, et al. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumors: a multicentre, single-group, phase 2 study. Lancet Oncol. 2015;16(6):695–703.
Xu J, Shen L, Zhou Z, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(11):1500–12.
Xu J, Shen L, Bai C, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(11):1489–99.
Capdevila J, Fazio N, Lopez C, et al. Lenvatinib in Patients With Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J Clin Oncol. 2021;39(20):2304–12.
Garcia-Carbonero R, Benavent M, Jiménez Fonseca P, et al. The AXINET trial (GETNE1107): Axitinib plus octreotide LAR improves PFS by blinded central radiological assessment vs placebo plus octreotide LAR in G1–2 extrapancreatic NETs. Ann Oncol. 2021;32(5):S906–20.
Bergsland EK, Mahoney MR, Asmis TR, et al. Prospective randomized phase II trial of pazopanib versus placebo in patients with progressive carcinoid tumors (CARC) (Alliance A021202). J Clin Oncol. 2019;37(15):4005–4005.
Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35:228.
Murat Cde B, da Rosa PW, Fortes MA, et al. Differential expression of genes encoding proteins of the HGF/MET system in insulinomas. Diabetol Metab Syndr. 2015;1(7):84.
Hansel DE, Rahman A, House M, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10(18 Pt 1):6152–8.
Peghini PL, Iwamoto M, Raffeld M, et al. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin Cancer Res. 2002;8(7):2273–85.
Krampitz GW, George BM, Willingham SB, et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc Natl Acad Sci U S A. 2016;113(16):4464–9.
Song J, Li M, Tretiakova M, et al. Expression patterns of PAX5, c-Met, and paxillin in neuroendocrine tumors of the lung. Arch Pathol Lab Med. 2010;134(11):1702–5.
Rai U, Thrimawithana TR, Valery C, Young SA. Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacol Ther. 2015;152:98–110.
Chalabi M, Duluc C, Caron P, et al. Somatostatin analogs: does pharmacology impact antitumor efficacy? Endocrinol Metab. 2014;25(3):115–27.
Pola S, Cattaneo MG. Vicentini LM Anti-migratory and anti-invasive effect of somatostatin in human neuroblastoma cells: involvement of Rac and MAP kinase activity. J Biol Chem. 2003;278(42):40601–6.
Azar R, Najib S, Lahlou H, et al. Phosphatidylinositol 3-kinase-dependent transcriptional silencing of the translational repressor 4E-BP1. Cell Mol Life Sci. 2008;65(19):3110–7.
Trusolino L, Bertotti A, Comoglio PM, et al. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–48.
Neuzillet C, de Mestier L, Rousseau B, et al. Unravelling the pharmacologic opportunities and future directions for targeted therapies in gastro-intestinal cancers part 2: Neuroendocrine tumours, hepatocellular carcinoma, and gastro-intestinal stromal tumours. Pharmacol Ther. 2018;181:49–75.
Ferone D, van Hagen PM, Semino C, et al. Somatostatin receptor distribution and function in immune system. Dig Liver Dis. 2004;36(Suppl 1):S68-77.
Levite M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr Opin Pharmacol. 2008;8(4):460–71.
Kwilas AR, Ardiani A, Donahue RN, Aftab DT, Hodge JW. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J Transl Med. 2014;13(12):294.
Lin YY, Tan CT, Chen CW, et al. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin Liver Dis. 2018;38(4):379–88.
Borghese Apolo A, Tomita Y, Lee MJ, et al. Effect of cabozantinib on immunosuppressive subsets in metastatic urothelial carcinoma. J Clin Oncol. 2014;32(15):4501–4501.
Duda DG, Zieh DR, Guo H, et al. Effect of cabozantinib treatment on circulating immune cell populations in patients with metastatic triple-negative breast cancer (TNBC). J Clin Oncol. 2016;34(15):1093–1093.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;42(2):228–47.
Krenning EP, Valkema R, Kooij PP, et al. Scintigraphy and radionuclide therapy with [indium-111-labelled-diethyl triamine penta-acetic acid-D-Phe1]-octreotide. Ital J Gastroenterol Hepatol. 1999;31(Suppl 2):S219–23.
Volante M, Brizzi MP, Faggiano A, et al. Somatostatin receptor type 2A immunohistochemistry in neuroendocrine tumors: a proposal of scoring system correlated with somatostatin receptor scintigraphy. Mod Pathol. 2007;20(11):1172–82.
Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10(1):1–10.
Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1814–23.
Choueiri TK, Halabi S, Sanford BL, et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol. 2017;35(6):591–7.
Elisei R, Schlumberger MJ, Müller SP, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31(29):3639–46.
Aboualfa GK, Meyer T, Cheng AL, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379(1):54–63.
Acknowledgements
This study was endorsed by the Italian Association for Neuroendocrine Tumors (It.a.net).
D issemination policy
The trial results will be presented at national and international conferences. Results will be published on top tier medical oncology journals. All Principal Investigators will be involved in manuscript writing.
Funding
The present study is an investigator-driven trial, carried out by participating clinicians, who have the intellectual ownership of the results. The study is sponsored by Fondazione IRCCS Istituto Nazionale dei Tumori, Via Venezian 1, 20133, Milan, Italy, that will provide the economical support for costs related to data management, statistical analysis and the other activities of central and group coordinating centers. Ipsen will provide study drugs (cabozantinib and lanreotide) and partial financial support for study costs. The study is endorsed by It.a.NET association.
The funders contributed and reviewed study design and reviewed the manuscript for accuracy but had no influence on the decision to publish.
Author information
Authors and Affiliations
Contributions
SP and LP: study design and conception; SP, LP, FC, NP, MM: writing of the original protocol; FC, SP, GR: writing of the original draft, review/editing and approval of the final manuscript; MPB, VA, DG, FP, DC, NP, MM, TC, CS, SO, GS, IP, BLS, LP, JC, MDB, FdB: review/editing and approval of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of the Fondazione IRCCS Istituto Nazionale dei Tumori di Milano (INT 240/19, v.1.1 21/10/2019) and by local Committees of participating Centers. The trial has been registered in EudraCT database (2019–004506-10) and at clinicaltrials.gov (NCT04427787). This study will be conducted in full conformance with the ICH E6/GCP guidelines and the principles of the Declaration of Helsinki. All study participants will provide their voluntary written informed consent before performance of any study-related procedure not part of normal medical care, with the understanding that consent may be withdrawn by the subject at any time without prejudice to future medical care. Any protocol amendments will be prepared by the Sponsor and submitted to the IRB/EC and to regulatory authorities in accordance with local regulatory requirements. If an amendment substantially alters the study design or increases the potential risk to the participant, patients will be re-consented to the most current version of the Consent Forms or to a significant addendum in accordance to IRB/IEC policy and applicable laws. The Sponsor maintains confidentiality standards by coding each patient through assignment of a unique patient identification number. Each participating Investigator is responsible for ensuring data quality. Patient medical information obtained by this study is confidential and may only be disclosed to third parties as permitted by the Informed Consent Form (or separate authorization for use and disclosure of personal health information) signed by the patient, unless permitted or required by law. Data generated by this study must be available for inspection upon request by representatives of the national and local health authorities, Sponsor’s monitors, representatives, and collaborators, and the IRB/EC for each study site, as appropriate. The Sponsor provides an insurance policy in case patients suffer harm from trial participation.
Consent for publication
Not applicable.
Competing interests
Dr. Pusceddu reported receiving honoraria from Novartis, Ipsen, Pfizer, Merck Serono, and Advanced Accelerator Applications (AAA); receiving grants from Ipsen and Pfizer; and receiving personal fees from AAA, Novartis, and Merck outside the submitted work. Dr. Prinzi reported receiving honoraria and travel accommodations from Novartis, Ipsen, Pfizer, Merck Serono, and Italfarmaco and receiving personal fees from Merck outside the submitted work. Dr. Panzuto reported receiving honoraria for speaker and advisory roles from Novartis, for an advisory role from Ipsen, and for consulting from AAA and receiving travel accommodations from Pfizer. Dr. Corti reported receiving nonfinancial support from AAA and Ipsen outside the submitted work. Dr. Porcu reported receiving honoraria for consulting from Ipsen and Italfarmaco. Dr. de Braud reported receiving honoraria for serving on advisory boards or speakers bureaus for Amgen, Novartis, Roche, Incyte, EMD Serono, Bristol Myers Squibb, Roche, Pfizer, Menarini, Sanofi, Healthcare Research & Pharmacoepidemiology, and Dephaforum; receiving research funding from Novartis, Roche, Merck Sharp & Dohme, Merck Serono, Pfizer, Servier, and Nerviano Medical Sciences; and receiving honoraria and/or personal fees from Roche, EMD Serono, Nerviano Medical Sciences, Sanofi, MSD, Novartis, Incyte, Bristol Myers Squibb, Menarini, Merck, Pfizer, Healthcare Research & Pharmacoloepidemiology, Servier, Amgen, and Dephaforum outside the submitted work. No other disclosures were reported.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Corti, F., Brizzi, M.P., Amoroso, V. et al. Assessing the safety and activity of cabozantinib combined with lanreotide in gastroenteropancreatic and thoracic neuroendocrine tumors: rationale and protocol of the phase II LOLA trial. BMC Cancer 23, 908 (2023). https://doi.org/10.1186/s12885-023-11287-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11287-2