
Abstract
Background
In this phase Ib/II open-label study, tumor immune suppression was targeted in patients with advanced refractory solid tumors and patients with recurrent/refractory non-small cell lung cancer (NSCLC) using galunisertib with nivolumab.
Methods
Eligible patients were ≥ 18 years old, had an Eastern Cooperative Oncology Group performance status ≤ 1, and were treatment-naive for anti-programmed cell death-1, its ligand, or transforming growth factor β receptor 1 kinase inhibitors. Phase Ib was an open-label, dose-escalation assessment of the safety and tolerability of galunisertib with nivolumab in patients with advanced refractory solid tumors. Phase II evaluated the safety of galunisertib with nivolumab in NSCLC patients who had received prior platinum-based treatment but were immuno-oncology agent-naive.
Results
This trial was conducted between October 2015 and August 2020. No dose-limiting toxicities were observed in phase I. In the phase II NSCLC cohort (n = 25), patients received 150 mg twice daily galunisertib (14 days on/14 days off dosing schedule for all phases) plus nivolumab at 3 mg/kg (intravenously every 2 weeks). In this phase, the most frequent treatment-related adverse events (AEs) were pruritus (n = 9, 36%), fatigue (n = 8, 32%), and decreased appetite (n = 7, 28%). No grade 4 or 5 treatment-related AEs were observed. Six (24%) patients had confirmed partial response (PR) and 4 (16%) had stable disease; 1 additional patient had confirmed PR after initial pseudo-progression. The median duration of response was 7.43 months (95% confidence interval [CI]: 3.75, NR). Among the 7 responders, including the delayed responder, 1 had high PD-L1 expression (≥ 50%). The median progression-free survival was 5.26 months (95% CI: 1.77, 9.20) and the median overall survival was 11.99 months (95% CI: 8.15, NR). Interferon gamma response genes were induced post-treatment and cell adhesion genes were repressed, although the association of these observations with tumor response and clinical outcomes was not statistically powered due to limited samples available.
Conclusions
The study met its primary endpoint as galunisertib combined with nivolumab was well tolerated. Preliminary efficacy was observed in a subset of patients in the Phase 2 NSCLC cohort.
Trial registration
Trial registered with ClinicalTrials.gov (NCT02423343; 22.04.2015).
Similar content being viewed by others

Background
Non-small cell lung cancer (NSCLC) accounts for approximately 85% of the incidences of lung cancer [1, 2]. Many tumors, including NSCLC, progress in part due to the acquisition of traits that allow cancer cells to evade immunosurveillance and escape the immune response.
Cancer immunotherapy aims to activate the host’s immune system to recognize and attack cancer cells. Recent successes in immunotherapy have been achieved by targeting immune checkpoints, i.e. a host of inhibitory receptors expressed on immune cells that upon activation suppress the inflammatory response [3]. Immune checkpoint inhibitors can induce durable responses and have demonstrated a survival benefit in patients with cancer, including metastatic, locally advanced, and early stage NSCLC [4,5,6,7].
Programmed cell-death-1 (PD-1) is expressed on activated T-cells and can act to dampen the immune response [8]. Tumor cells overexpress PD-1 ligand (PD-L1), either by pro-inflammatory stimuli or as a result of pro-oncogenic pathway activation, and inhibit the local immune response [9]. Nivolumab blocks the binding of PD-L1 and PD-L2 to its receptor, allowing the activated T-cells to identify and attack cancer cells [10]. Although NSCLC may have a high mutational load or PD-L1 expression, which are associated with response to anti-PD(L)1 therapy, many patients do not respond to single agent anti-PD(L)1 inhibition [11, 12].
Transforming growth factor β (TGF-β) signaling plays an important role in tumorigenesis and contributes to many hallmarks of cancer cells including cell proliferation, invasion, escape of immune surveillance, angiogenesis, and metastasis [13]. A major contributing factor for mortality in NSCLC, like most cancer types, is metastasis [14]. Epithelial-mesenchymal transition (EMT) is a crucial event leading to metastasis during which cells exhibit mesenchymal properties such as becoming more motile and invasive [15]. TGF-β signaling is a primary inducer of EMT in NSCLC [16]. TGF-β binding to its receptor leads to phosphorylation of small mothers against decapentaplegic homolog 2 (SMAD2) and SMAD3, which then form a transcriptional complex with SMAD4, activating the expression of target genes [17]. SMAD2 has been identified as a key element downstream of the TGF-β signaling pathway in regulating cancer metastasis through promoting EMT [18, 19]. Upregulation of SMAD2 has been associated with poor survival in patients with NSCLC [20].
Galunisertib is an oral small molecule inhibitor of the TGF-β receptor 1 kinase that specifically down-regulates the phosphorylation of SMAD2 and is associated with an increase in T-cell infiltration in tumors [21, 22].
TGF-β can suppress or alter the activation, maturation, and differentiation of both innate and adaptive immune cells [23]. Increased TGF-β in the tumor microenvironment promotes T-cell exclusion from tumors, and blocks acquisition of the T helper cells-effector phenotype, which are both associated with poor clinical outcomes. Inhibition of TGF-β releases a cytotoxic T-cell response against tumor cells and allows immune cells to infiltrate the tumor [24]. However, inhibition of TGF-β on its own is not always sufficient to promote tumor rejection [23]. In preclinical models, concurrent blockade of TGF-β and PD-L1 work synergistically to reverse immunosuppression leading to T-cell infiltration and activation, which promote antitumor activity [24,25,26].
In this study, both TGF-β and PD-1 were targeted using galunisertib in combination with nivolumab in patients with advanced refractory solid tumors and in patients with recurrent/refractory NSCLC who were PD-(L)1 naïve. The clinical trial objectives were to evaluate the safety and tolerability of the drug combination.
Patients and methods
Study design
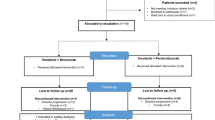
This was a phase Ib/II open-label study (NCT02423343) conducted between October 2015 and August 2020. The phase Ib portion of this study consisted of a dose-escalation assessment of the safety and tolerability of galunisertib administered at 50 mg daily, 50 mg twice daily (BID), 80 mg BID, or 150 mg BID in combination with nivolumab 3 mg/kg administered every two weeks in patients with advanced refractory solid tumors. The phase II portion of the trial evaluated the safety and efficacy of 150 mg BID galunisertib in combination with nivolumab at 3 mg/kg in patients with recurrent or refractory NSCLC that were immune checkpoint naive. The study did not have a fixed treatment duration, and patients received combination treatment in 28-day cycles until disease progression, intolerable toxicity or withdrawal of consent. Based on the results of a preclinical dosing schedule investigation that evaluated toxicity using animal models [27], each patient took galunisertib on an intermittent dosing strategy of a 14 days on/14 days off dosing schedule for all phases of the trial.
Eligibility criteria
Inclusion and exclusion criteria for phase Ib and phase II are shown in Additional file 1. In the phase 1a escalation portion, patients with any solid tumor in the advanced stage refractory to standard of treatment were eligible. The phase II portion of this study included patients with stage IV NSCLC who were previously treated with platinum-based chemotherapy. Patients with actionable oncogene mutations were required to receive prior treatment with tyrosine kinase inhibitor (TKI) (e.g., approved EGFR TKI for patients with activating EGFR mutations and approved ALK TKI for patients harboring ALK-fusions). Patients that received more than 1 line of treatment in the advanced setting were excluded. Eligible patients were ≥ 18 years old with an Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1, were treatment-naive for anti-PD-(L)1 or TGF-β receptor 1 kinase inhibitors and had measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 [28].
Efficacy measures
Efficacy measures included overall survival (OS), progression-free survival (PFS), overall response rate [ORR -partial response (PR) + complete response (CR)] and duration of response (DoR). Tumor responses were assessed per RECIST v1.1 guidelines. DoR, PFS and OS were estimated using Kaplan- Meier methodology.
Outcomes
The primary objective of the study (both phases) was to assess the safety and tolerability of galunisertib in combination with nivolumab by identifying dose-limiting toxicities (DLTs) and the maximum tolerated dose or pharmacologically active dose of the combination in patients with advanced refractory or solid tumors during the first 2 cycles. DLTs were defined as Grade 3 non-haematologic toxicity; Grade 4 haematological toxicity of > 5 days duration; any febrile neutropenia.
The secondary objectives of the study were to characterize the pharmacokinetics (PK) of galunisertib and nivolumab when co-administered, to characterize the immunogenicity of nivolumab when administered in combination with galunisertib, and to estimate the OS rate. In addition, PFS, ORR, and DOR were evaluated for patients with NSCLC in phase II. Exploratory objectives were to examine biomarkers and correlate these makers to clinical outcomes.
Toxicity and safety measures
All patients who received at least 1 dose of either galunisertib or nivolumab were evaluated for safety and toxicity. Any other significant toxicity deemed to be dose limiting or resulted in the patient getting < 75% of the total doses or lead to holding galunisertib for > 2 weeks. For nivolumab, any toxicity that occurred during Cycles 1 or 2 managed by discontinuation. Any Grade ≥ 2 nonskin, drug-related AE; any Grade 3 skin, drug related AE; any Grade 3 drug-related laboratory abnormality; any adverse event (AE), laboratory abnormality, or intercurrent illness which, in the judgment of the investigator, warranted delaying the dose of study medication. Exceptions not considered a DLT included: Grade 3 amylase or lipase abnormalities not associated with symptoms or clinical manifestations of pancreatitis that did not require a dose delay; nausea, vomiting, diarrhea, and constipation controlled with treatment; fatigue relieved by rest; Grade 3 elevations of alanine aminotransferase and/or aspartate aminotransferase, without evidence of other hepatic injuries; anorexia not associated with significant weight loss (weight loss of > 10% baseline) or malnutrition. The AEs were assessed, according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0, continuously during the study and for 100 days after the last dose of combination treatment. Disease assessment with computed tomography and/or magnetic resonance imaging, as appropriate, were performed at baseline and approximately between Days 22 and 28 of every other cycle and completed before the first dose in the next cycle, until disease progression or patient withdrawal from the study.
Pharmacokinetics and biomarkers
PK of galunisertib were measured using validated liquid chromatography-atmospheric pressure ionization/tandem mass spectrometry methods at Intertek Pharmaceutical Services (El Dorado Hills and San Diego, California, USA). There were 293 PK galunisertib observations in 41 patients on Days 1, 14, and 15 in Cycles 1 and 2 as well as pre-dose, Cycle 4 Day 1 (C1D4).
Tumor cell membrane PD-L1 was measured by immunohistochemistry using the Dako PD-L1 (28–8) kit at Mosaic Laboratories (n = 20 samples) and scored as percent tumor cells expressing PD-L1. Anti-drug antibodies (ADA) were evaluated in all patients comparing their baseline levels to study levels. Patients that developed positive ADA after negative baseline were further evaluated for AEs and response to treatment to assess potential relationship between study treatment and immunogenicity.
Cancer gene sequencing (n = 9 tumor samples) was performed by Foundation Medicine using the FoundationOne T7 assay (404 genes). Here we report results for tumor mutation burden (TMB) as mutations/megabase and genetic variants for common mutated genes or genes with known relevance to anti-PD(L)1 NSCLC response.
Ribonucleic acid (RNA) extraction and Illumina TruSeq RNA Exome sequencing was performed by Almac for gene expression profiling of 12 baseline tumor samples, 4 tumors collected on C1D15, and 3 tumors collected on C2D1. Six baseline samples were paired with treatment biopsies. Formalin fixed paraffin embedded tumor samples were macrodissected and RNA extraction was performed using the Qiagen RNeasy extraction kit followed by quality assessments using Nanodrop, Agilent Bioanalyzer & RNA-Seqability (an Almac proprietary quality control step). Paired-end sequencing with a read length of 100 base pairs and targeted read depth of 50 million reads/sample was performed. Read counts were quantified using an in-house Perl script and summarized at the gene level (National Center for Biotechnology Information [NCBI] h37.p13 annotation). The resulting data were quantile-normalized and filtered to remove genes with fewer than 5 counts across 80% of the samples from the analysis. Baseline expression of TGF-β pathway genes included TGFβ-1, TGFβ-2, ACVR1, TGFβ-3, TGF-β R1, TGF-β R2, SMAD2, SMAD4; and T-cell inflamed genes included IFNGR1, IFNG, TIGIT, CD27, CD8A, PDCD1LG2, LAG3, CD274, CXCR6, CMKLR1, NKG7, CCL5, PSMB10, IDO1, CXCL9, HLA-DQA1, CD276, STAT1, HLA-DRB1, and HLA-E.
Differentially expressed gene analysis was conducted using the DESeq2 package [29]. Fold changes were calculated to show up or down-regulation of genes between 4 tumors collected on C1D15 and 3 tumors collected on C2D1 versus 12 baseline tumor samples. Differentially expressed genes were identified from comparisons when the p-value was ≤ 0.05 and the absolute fold change was ≥ 1.5. The contrasts of differentially expressed genes were displayed in a heatmap using the Complex Heatmap R package (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html).
Serum proteins (51 analytes) were assessed at baseline, C1D8, C1D15, C2D1, and C2D15 using the Inflammation Multi-Analyte Immunoassay panel developed by Myriad RBM. Paired t-tests were used to compare post-dose levels with baseline at each time point for each biomarker.
Statistical analysis
All patients who received any dose of study treatment were included in safety and efficacy analyses. For safety analyses, the frequency and percentage of patients with dose-limiting toxicities and AEs were presented for each cohort. Best overall response per RECIST v1.1 with confirmation on CR and PR were represented by frequency, percentage, and 95% confidence interval (CI) with the Clopper-Pearson Confidence Interval method. PFS, DOR, and OS were analyzed using the Kaplan–Meier method. PFS was defined as the time from the date of first study treatment to the first date of documented progression or death due to any cause. For patients who were not known to have died or progressed as of the cut-off dates, PFS times were censored at the date of the last progression-free disease assessment prior to the date of any subsequent anticancer therapy. DOR was measured from the date of the first documented response to the date of first disease progression or the date of death due to any cause, using the same censoring rules as PFS. OS was defined as the time from the date of first study treatment to the date of death from any cause. For each patient who was not known to have died as of the cut-off date, OS data were censored for that analysis at the date of last contact prior to the data inclusion cutoff date. SAS 9.4 was used for analyses on the clinical data for demographics, baseline characteristics, safety, and efficacy.
Results
Baseline patient demographics
Fifteen patients were enrolled in phase Ib and 25 patients in phase II. Baseline patient and disease characteristics of patients in phase 1b are depicted in Additional file 2. In the phase II NSCLC cohort, 16 (65%) of the patients were male and all were Caucasian. Two (8%) had tumors with PD-L1 expression of ≥ 50%, 11 (44%) had PD-L1 expression ranging from 1–49%, and 7 (28%) had no PD-L1 expression. Demographic and baseline disease characteristics of the patient population are shown in Table 1.
Toxicity and safety
The primary objective of safety was reached for both phases of the trial. No DLTs were observed in the phase Ib portion of the study. The median duration of galunisertib treatment ranged from 56 days (min 28 days; max 157 days) to 143 days (min 55 days; max 462 days) across the phase Ib cohorts. The most common AEs related to study treatment in phase Ib were observed in the galunisertib 150 mg BID cohort and included rash maculo-papular, amylase increased, aspartate aminotransferase increased, gamma-glutamyltransferase increased, blood alkaline phosphatase increased, lipase increased, and non-cardiac chest pain. The median duration of galunisertib treatment was 120 days (min 14 days; max 726 days) in the phase II NSCLC cohort. Two deaths occurred on treatment (multi-organ failure and myocardial infarction) in phase II. Both cases were deemed unrelated to study treatment by the treating physicians. The case of multi-organ failure was deemed related to study disease and, the case of myocardial infarction was considered unrelated as patient had pre-existing underlying conditions that increased the risk of cardio-vascular disease, and a familial history of cardiac disease. There were no Grade 4 or 5 treatment-related toxicities. The most frequent treatment-related AEs were pruritus (n = 8, 32%), fatigue (n = 8, 32%), and decreased appetite (n = 7, 28%). Other treatment-related Grade 3 AEs included immune-related encephalitis, diarrhea, fatigue, aspartate aminotransferase/alanine aminotransferase/gamma-glutamyltransferase increase, blood alkaline phosphatase increase, abdominal distension, cutaneous rash (n = 1 each), and cholestasis (n = 2) that resolved or were resolving at the time of data cutoff (Table 2). A total of 28 patients (68%) across the phase Ib cohorts and the phase II NSCLC cohort experienced a Grade ≥ 3 AE, of which 12 (29%) were related to treatment. Discontinuation rate due to any treatment related AE was 2%.
Efficacy
No patients in the phase Ib portion of the study achieved CR or PR, though stable disease (SD) was observed in seven patients across the four dosing schedules. In the phase II NSCLC portion of the trial, 6 (24%) patients had PR, no patient achieved CR, 4 (16%) had stable disease (SD), 9 (36%) patients had progressive disease (PD), 1 of 9 progressed patients had confirmed PR after initial pseudo-progression (delayed responder), and 6 (24%) patients were deemed not evaluable (NE) due to treatment discontinuation (Fig. 1). Reasons for treatment discontinuation included AEs, subject decision, and PD. The median DOR was 7.41 months (95% CI: 3.75, not reached [NR]), and the median PFS was 5.26 months (95% CI: 1.77, 9.20) (Fig. 2). The median OS was 11.99 months (95% CI: 8.15, NR).

Waterfall plot of best overall response rate (ORR) according to RECIST in the in NSCLC Cohort. Information is provided about baseline PD-L1 expression in 18 patients (number in blue), and tumor mutation burden (TMB) in 7 patients (number in red). Abbreviations: NSCLC, non-small cell lung cancer; PD-L1, programmed death ligand 1

Efficacy outcomes in the phase II NSCLC cohort. Kaplan–Meier plots of Progression-Free Survival (A) Overall Survival (B) and Duration of Response (C)
Pharmacokinetics
Phase Ib PK data showed rapid absorption (1–3 h) and elimination of galunisertib within 48 h. Observed galunisertib plasma concentrations were comparable with those observed in previous galunisertib trials [30].
There was no significant immunogenicity observed that would have impacted study results.
Biomarkers
Tumor PD-L1 expression was measured by immunohistochemistry. Among 7 responders, including the delayed responder, only 1 patient had high PD-L1 expression (≥ 50%), 5 patients had PD-L1 scores in the range of 1 to 30%, and 1 patient was PD-L1 negative. PD-L1 expression did not correlate with efficacy based upon this small sample set (Fig. 1).
DNA sequencing was performed for the subset of tumor samples with sufficient tissue remaining (N = 9) (Fig. 3). As expected, the TP53 gene had the greatest frequency of pathogenic variants detected (8 of 9, 89%). Two of the samples associated with PR harbored variants in CDKN2A and 2 other samples with NFE2L2 variants were both associated with PD. No STK11 mutant tumors were represented in the subset of samples with genetic data, but 2 KEAP1 mutated tumors were observed in patients with a PR (Fig. 3). This data set was not powered to correlate individual gene alterations or TMB with clinical response.

Genetic Variants and RNA Signatures in NSCLC Cohort. Abbreviations: BOR, best overall response; CTS, change in tumor size; NE, not evaluable; OS, overall survival; PD, progressive disease; PD-L1, programmed death ligand 1; PFS, progression-free survival; PR, partial response; TGF-β, transforming growth factor beta; TMB, tumor mutation burden. *pseudo-progressor patient
Baseline RNA expression data associated with 12 tumor samples was used to evaluate previously identified expression signatures associated with immune function and TGF-β biology, including the T-cell inflamed signature and TGF-β pathway genes [31]. Within this small data set elevated expression of TGF-β pathway and T-cell inflamed status tended to be aligned with tumor regression and survival (Fig. 3).
The RNA expression profile of 19 tumor samples were analyzed (12 baseline samples vs 4 collected on C1D15 and 3 on C2D1) to look for pharmacodynamics (treatment driven) expression changes. Of the 6 paired samples, 3 were baseline versus C1D15 and 3 were baseline versus C2D1. Genes associated with interferon gamma response were upregulated, while repressed genes were associated with cell adhesion (Fig. 4). TGF-β pathway genes were not modulated (data not shown). The genes with the largest fold induction at C1D15 were the chemokines CXCL10, CXCL9, and CXC11, which play a role in attracting immune cells, such as cytotoxic T-cells, natural killer (NK) cells, and macrophages. Only CXCL9 was induced at C2D1 (unadjusted p-value < 0.05). These changes were not associated with response as the paired samples were collected from no-responders.

Modulation of Tumor RNA Expression in NSCLC Cohort. Abbreviations: CxDx, Cycle x Day x; FC, fold change; IFNG.RES, interferon gamma response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease. Shapes represent 6 paired samples; the star represents the pseudo-progressor patient; filtering criteria: p ≤ 0.05 and |FC|≥ 1.5
Serum proteins (n = 51) were assessed at baseline and at specific time points during the dosing period. No significant association between baseline protein levels and PFS, OS, or response were detected after adjusting for multiplicity (n = 19 patients). Analysis of serum protein modulation in response to treatment (PK), also did not detect changes significant after correction for multiplicity, however, a trend of increased interferon gamma-induced protein 10 (IP-10) (CXCL10), monokine induced by gamma interferon (MIG) (CXCL9), and interleukin-2 receptor alpha was observed on treatment (Fig. 5). The increase in circulating IP-10 and MIG is consistent with the observed increase in RNA expression of their encoding genes CXCL10 and CXCL9 in tumor samples.

Serum Interferon Gamma Induced Protein 10, Interleukin-2 Receptor Alpha, and Monokine Induced by Gamma Interferon in NSCLC Cohort. Abbreviations: NSCLC, non-small cell lung cancer; CxDx, Cycle X Day X; IP-10, interferon gamma-induced protein 10; MIG, monokine induced by gamma interferon
Discussion
Immune checkpoint molecules are established targets for cancer immunotherapies. Blocking PD-(L)1 signaling has become standard of care in patients with lung cancer in different clinical settings, but still not all patients benefit from these therapeutic approaches [32]. Several studies have demonstrated the benefit of anti-PD-(L)1 in combination with chemotherapy and studies are now investigating whether combined blockade of PD-(L)1 and TGF-β signaling can induce tumor regression [33,34,35]. TGF-β signaling plays an important role in tumorigenesis by inducing EMT. EMT is a major contributing factor of mortality in a range of malignancies including NSCLC, since it is associated with cancer progression and metastasis [13,14,15, 36].
Overall, in this study we did not observe significant PFS or OS benefit suggesting a delay in disease progression, although some patients that had elevated baseline expression of TGF-β pathway genes tended to have longer PFS and OS benefits than patients who did not (Fig. 3).
Given the immune suppressive role of TGF-β on cytotoxic T-cells, our clinical hypothesis was that dual blockade of both TGF-β and PD-1 may overcome the immunosuppressive nature of the tumor microenvironment and lead to T-cell infiltration and activation [37]. In our study, we observed the combination treatment of galunisertib at the recommended phase II dose of 150 mg BID for 14 days on/14 days off with nivolumab 3 mg/kg every 2 weeks to be well tolerated. No DLTs were observed in the phase Ib portion of the study and the safety profile of the combination was not different than the safety profiles of nivolumab or other checkpoint inhibitors. The most commonly reported treatment-related AEs were pruritus, fatigue, and decreased appetite. Except for 1 myocardial infarction leading to death, deemed unrelated to study treatment due to underlying comorbidities and a familial history of cardiac disease in the patient, no cardiovascular toxicities were observed, which was a major concern from preclinical toxicology studies.
The 2-week treatment break and a maximum dose of 150 mg BID was introduced not to exceed area under the curve (AUC) levels corresponding to levels where valvulopathies were observed in animals. Although we did not observe major cardiovascular signals or emergence of secondary malignancies in our study, it is plausible higher doses might have led to better target inhibition and thus higher response rates. Interestingly, TGF-β pathway genes were not modulated but changes were seen in interferon gamma gene expression that is likely related to PD-1 blockade. Although most patients had tumor target reduction, overall, the efficacy data were not different than PD-(L)1 monotherapy in the same disease setting.
The median OS was 11.99 months (95% CI: 8.15, NR) for patients treated with galunisertib plus nivolumab. This is not different than survival data observed in multiple large studies showing median OS to be between 12–14 month for anti PD-1 (nivolumab, pembrolizumab) and anti PD-L1 (durvalumab, atezolizumab) single agent treatment [4, 38,39,40]. Here, we report the median PFS to be 5.26 months. This compares favorably to single agent PD-(L)1 trials where median PFS was between 2.8 to 3.8 months depending on the trial [4, 5]. However, given the small sample size and the nonrandomized design of this trial, it is not possible to draw any conclusions as to whether the addition of the TGF-β inhibitor provided any clinical benefit. Studies have shown variable associations between PD-L1 expression and response to nivolumab in NSCLC [4, 41,42,43]. Here, we did not observe clear associations between response and PD-L1 expression due to the limited sample size, however most patients with PD-L1 positivity had tumor reduction (Fig. 3).
DNA sequencing was performed to assess for associations between genetic variants and clinical response. Both STK11 and KEAP1 mutations have been associated with resistance to checkpoint blockade, especially when concurrent with kirsten rat sarcoma viral oncogene homolog (KRAS) mutations [44, 45]. None of the patients that had tumor tissue available for sequencing had STK11 mutations suggesting loss of liver kinase B1. Two patients harbored a KEAP1 mutation, which has been associated with tumor progression and treatment resistance in lung cancer [46, 47]. Interestingly, both patients with a KEAP1 mutation achieved a PR, despite the association with poor response to checkpoint inhibitors in combination with chemotherapy in first line therapy [45]. Two samples had a KRAS mutation. One KRAS (G12C) variant without co-occurring mutations in TP53 and STK11 achieved a PR, while the other with KRAS (G12S) variant concurrent with a TP53 mutation progressed on treatment. An ARID1A mutation was found in 1 patient sample, which has been associated with a better response to checkpoint inhibitors and the patient response observed here was a PR [48, 49]. Finally, TMB is a surrogate of tumor antigenicity and has been positively associated with a response to nivolumab and other checkpoint inhibitors [12, 50, 51]. In our study, no association of TMB score with response was observed, however, data are insufficient to make any conclusions.
Patients were not selected prospectively based on TGF-β gene expression, therefore the appropriate patient population may not have been treated to see changes in the expression of TGF-β pathway genes. Higher doses may have modulated gene expression but were associated with higher risk of inducing cardiac toxicity. Notwithstanding, we detected increased transcription and serum levels of T-cell activation and migration markers IP-10 (CXCL10) and MIG (CXCL9) following combination treatment of nivolumab and galunisertib. Similarly, increased transcription and serum levels of these same markers were reported in renal cell carcinoma patients post nivolumab treatment [52]. These changes may be indicative of treatment driven T-cell recruitment to the tumor microenvironment.
These findings demonstrate combination treatment with galunisertib plus nivolumab has an acceptable safety profile in patients with refractory or recurrent NSCLC who received prior platinum-based treatment and were treatment-naive for anti-PD-(L)1 or TGF-β receptor 1 kinase inhibitors. Notwithstanding, this study has several limiting factors. The primary limitation is the low number of patients. While this study contributes to research in the field, analysis of a larger cohort of patients would provide further data on the safety and efficacy of the combination treatment in NSCLC. Another limitation is the low number of paired baseline and on-treatment tumor samples for exploratory comparative analysis. Collection of baseline and on-treatment biopsies were required for participation in this study, however, limited on-treatment tissue was submitted due to tumor progression, clinical deterioration, insufficient tumor, and patient decision. Thus, although these results identify potential predictive markers of therapeutic effect in recurrent or refractory NSCLC, more patient samples are needed to support the translational work.
In conclusion, combination treatment of galunisertib with nivolumab was well tolerated and the study met its primary endpoint of safety. Preliminary efficacy activity was observed in a subset of patients and was not associated with tumor PD-L1 expression. Increased levels of T-cell activation and migration markers were indicative of potential treatment-driven T-cell recruitment. However, the sample size was small and the magnitude of response or benefit in survival outcomes do not clearly differentiate from nivolumab monotherapy in the second line IO-naïve setting. In this unselected cohort of NSCLC patients, a few patients exhibited a baseline increase in TGF-β activity that may have benefited from this combination treatment. Further studies that select patients based on TGF-β pathway expression may yield higher response rates to TGF-β and immune checkpoint inhibitor combination treatment.
Availability of data and materials
The data that support the findings of this study are available from Eli Lilly and Company but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors (Emin Avsar, Ivelina Gueorguieva, Michael Man, Shawn T Estrem, Jiangang Liu) upon reasonable request and with permission of Eli Lilly and Company.
Abbreviations
- AE:
-
Adverse event
- ARID1A:
-
AT-rich interaction domain 1A
- AUC:
-
Area under the curve
- BID:
-
Twice daily
- BOR:
-
Best overall response
- CDKN2A:
-
Cyclin dependent kinase inhibitor 2A
- CI:
-
Confidence interval
- CR:
-
Complete response
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- CTS:
-
Change in tumor size
- CXCL:
-
C-X-C motif chemokine ligand
- CxDy:
-
Cycle x Day y
- DLT:
-
Dose-limiting toxicity
- DOR:
-
Duration of response
- ECOG:
-
Eastern Cooperative Oncology Group
- EMT:
-
Epithelial-mesenchymal transition
- IP-10:
-
Interferon gamma-induced protein 10
- KEAP1:
-
Kelch-like ECH-associated protein 1
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- MIG:
-
Monokine induced by gamma interferon
- NE:
-
Not evaluable
- NFE2L2:
-
Nuclear factor, erythroid 2 like 2
- NK:
-
Natural killer
- NR:
-
Not reached
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PD:
-
Progressive disease
- PD-1:
-
Programmed cell-death-1
- PD-L1:
-
PD-1 ligand
- PFS:
-
Progression-free survival
- PK:
-
Pharmacokinetics
- PR:
-
Partial response
- RECIST:
-
Response evaluation criteria in solid tumors
- RNA:
-
Ribonucleic acid
- SD:
-
Stable disease
- SMAD2/3/4:
-
Mothers against decapentaplegic homolog 2/3/4
- STK11:
-
Serine/threonine kinase 11
- TGF-β:
-
Transforming growth factor β
- TKI:
-
Tyrosine kinase inhibitor
- TMB:
-
Tumor mutation burden
- TP53 gene:
-
Tumor protein p53
References
Society AC. Cancer Facts & Figures 2019 [Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54.
Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–61.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35.
Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, et al. Neoadjuvant Nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386(21):1973–85.
Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet. 2021;398(10308):1344–57.
Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32(3):634–43.
Escors D, Gato-Cañas M, Zuazo M, Arasanz H, García-Granda MJ, Vera R, et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct Target Ther. 2018;3:26.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30.
Galuppini F, Dal Pozzo CA, Deckert J, Loupakis F, Fassan M, Baffa R. Tumor mutation burden: from comprehensive mutational screening to the clinic. Cancer Cell Int. 2019;19(1):209.
Huang JJ, Blobe GC. Dichotomous roles of TGF-β in human cancer. Biochem Soc Trans. 2016;44(5):1441–54.
Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–95.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Yang H, Zhan L, Yang T, Wang L, Li C, Zhao J, et al. Ski prevents TGF-β-induced EMT and cell invasion by repressing SMAD-dependent signaling in non-small cell lung cancer. Oncol Rep. 2015;34(1):87–94.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179(1–2):11–23.
Chen T, Zhu J, Cai T, Du W, Zhang Y, Zhu Q, et al. Suppression of non-small cell lung cancer migration and invasion by hsa-miR-486-5p via the TGF-β/SMAD2 signaling pathway. J Cancer. 2019;10(24):6014–24.
Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479–99.
Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer. 2018;6(1):47.
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–43.
Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFβ blockade augments PD-1 inhibition to promote t-cell-mediated regression of pancreatic cancer. Mol Cancer Ther. 2019;18(3):613–20.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8.
Stauber A, Credille K, Truex LL, Ehlhardt W, Young J. Nonclinical safety evaluation of a transforming growth factor β receptor I kinase inhibitor in Fischer 344 rats and beagle dogs. J Clin Toxicol. 2014;4:3.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Gueorguieva I, Tabernero J, Melisi D, Macarulla T, Merz V, Waterhouse TH, et al. Population pharmacokinetics and exposure-overall survival analysis of the transforming growth factor-β inhibitor galunisertib in patients with pancreatic cancer. Cancer Chemother Pharmacol. 2019;84(5):1003–15.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–40.
Grigg C, Rizvi NA. PD-L1 biomarker testing for non-small cell lung cancer: truth or fiction? J Immunother Cancer. 2016;4(1):48.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–51.
Doi T, Fujiwara Y, Koyama T, Ikeda M, Helwig C, Watanabe M, et al. Phase I study of the bifunctional fusion protein Bintrafusp Alfa in Asian patients with advanced solid tumors, including a hepatocellular carcinoma safety-assessment cohort. Oncologist. 2020;25(9):e1292–302.
Yap T, Barve M, Gainor J, Bockorny B, Ju Y, Cote S, et al. 532 First-in-human phase 1 trial of SRK-181: a latent TGFβ1 inhibitor, alone or in combination with anti-PD-(L)1 treatment in patients with advanced solid tumors (DRAGON trial). J. Immunother. Cancer. 2021;9(Suppl 2);A563-A.
Wang H, Wu Q, Zhang Y, Zhang HN, Wang YB, Wang W. TGF-β1-induced epithelial-mesenchymal transition in lung cancer cells involves upregulation of miR-9 and downregulation of its target E-cadherin. Cell Mol Biol Lett. 2017;22:22.
Sow HS, Ren J, Camps M, Ossendorp F, Ten Dijke P. Combined inhibition of TGF-β signaling and the PD-L1 immune checkpoint is differentially effective in tumor models. Cells. 2019;8(4):320.
Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665–73.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–65.
Planchard D, Reinmuth N, Orlov S, Fischer JR, Sugawara S, Mandziuk S, et al. ARCTIC: durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann Oncol. 2020;31(5):609–18.
Rizvi NA, Shepherd FA, Antonia SJ, Brahmer JR, Chow LQ, Goldman J, et al. First-line monotherapy with Nivolumab (Anti-PD-1; BMS-936558, ONO-4538) in advanced Non-Small Cell Lung Cancer (NSCLC): safety, efficacy, and correlation of outcomes with PD-L1 status: metastatic non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2014;90(5 Supplement):S31.
Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall survival and long-term safety of Nivolumab (Anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol. 2015;33(18):2004–12.
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of Nivolumab in previously treated advanced non-small-cell lung cancer: results from the CA209-003 study. J Clin Oncol. 2018;36(17):1675–84.
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–35.
Papillon-Cavanagh S, Doshi P, Dobrin R, Szustakowski J, Walsh AM. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open. 2020;5(2):e000706.
Morén A, Raja E, Heldin CH, Moustakas A. Negative regulation of TGFβ signaling by the kinase LKB1 and the scaffolding protein LIP1. J Biol Chem. 2011;286(1):341–53.
Barrera-Rodríguez R. Importance of the Keap1-Nrf2 pathway in NSCLC: Is it a possible biomarker? Biomed Rep. 2018;9(5):375–82.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–62.
Rizvi N, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M, et al. OA04.07 mutations associated with sensitivity or resistance to immunotherapy in mNSCLC: analysis from the MYSTIC trial. J Thor Oncol. 2019;14(10):217.
Krieger T, Pearson I, Bell J, Doherty J, Robbins P. Targeted literature review on use of tumor mutational burden status and programmed cell death ligand 1 expression to predict outcomes of checkpoint inhibitor treatment. Diagn Pathol. 2020;15(1):6.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–104.
Choueiri TK, Fishman MN, Escudier B, McDermott DF, Drake CG, Kluger H, et al. Immunomodulatory activity of Nivolumab in metastatic renal cell carcinoma. Clin Cancer Res. 2016;22(22):5461–71.
Acknowledgements
We thank our patients and their caregivers for their participation in this study, the study investigators and their staff, the independent data monitoring committee, and the JBEF clinical trial team. This trial was conducted in collaboration with Bristol Myers Squibb, which supplied nivolumab. Sarah Beckman and Adrija Tripathy of Syneos Health and Declan O’Dea of Eli Lilly and Company provided medical writing and editorial support, funded by Eli Lilly and Company.
Funding
Funded by Eli Lilly and Company in collaboration with Bristol Myers Squibb.
Author information
Authors and Affiliations
Contributions
S. Guba, I. Ales Diaz, E. Nadal, S. Patel, M. Saleh, E. Avsar, and S. Estrem acquired data. Y. Zhao, M. Man, J. Liu, I. Gueorguieva, S. Estrem, E Avsar, M. Saleh, E. Nadal, L. Gandhi, and S. Guba analyzed and interpreted the data. S. Patel interpreted the data. S. Guba, E. Avsar, J. Liu, S. Estrem, M. Man, and Y. Zhao participated in drafting of the manuscript. All authors revised the work critically for important intellectual content. All authors made substantial contributions, gave final approval for the work to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol was approved by University of Texas MD Anderson Cancer Center, Western Institutional Review Board WIRB, Dana Farber Cancer Institute, Chesapeake Research Review Inc, University of California San Diego, and Hospital Universitari Vall d’Hebron. Local ethical review boards and all methods were conducted according to the International Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. Informed consent was obtained from all the participants.
Consent for publication
Not required.
Competing interests
This work was supported by Eli Lilly and Company. S. Estrem, I. Gueorguieva, M. Man, and J. Liu are full time employees and minor stockholders of Eli Lilly and Company. L. Ghandi was a full time employee of Eli Lilly and Company at the time of this work and is minor stockholder of Eli Lilly and Company, has received consultancy fees from Pilant Therapeutics, 28–7 Therapeutics, Riva Therapeutics, and Eli Lilly and company, has received speaker's bureau honoraria from Tempus, is on the advisory board for Beigene, Bristol-Myers Squibb, Mersana, Eisai, Xilio, and Tentarix, and is on the board of directors for Bright Peak Therapeutics an Neximmune. Y. Zhao, and S. Guba were full time employees and minor stockholders of Eli Lilly and Company at the time of this work. S. Guba has also received medical writing support from Eli Lilly and Company. K.A. Benhadji owns a patent with and is a minor stockholder of Eli Lilly and Company. E. Avsar is a full time employee of Eli Lilly and Company and a minor stockholder in Eli Lilly and Company and BMS, and was a full time employee of BMS at the time of this work. W. H Lin was an employee of BMS when this work was performed. E. Nadal has received grants or research funding from Bristol-Myers Squibb, Eli Lilly and Company, Pfizer, Roche, and Merck Serono, and has received speaker's bureau honoraria from Roche, Bristol Myers Squibb, Merck Sharp Dohme, Merck-Serono, Sanofi, Pfizer, Eli Lilly, Amgen, Boehringer-Ingelheim, AstraZeneca, Takeda, Sanofi, Janssen, Qiagen and Bayer. S. Patel is an advisory board member for Amgen, AstraZeneca, Bristol-Myers Squibb, Certis, Eli Lilly and Company, Jazz, Genentech, Illumina, Merck, Pfizer, Rakuten, and Tempus. S. Patel's university receives research funding from Amgen, AstraZeneca/MedImmune, Bristol-Myers Squibb, Eli Lilly and Company, Fate Therapeutics, Iovance, Merck, Pfizer, Roche/Genentech, and SQZ Biotechnologies. M. Saleh received medical writing support from Eli Lilly and Company. S. Antonia has been a consultant or advisory board member for Achilles Therapeutics, Amgen, Bristol-Myers Squibb, Celsius Therapeutics, G1 Therapeutics, Glimpse, Guardian Bio, GSK, Immutep, Memgen, Merck, RAPT Therapeutics, Shoreline, Taurus, Xilis, has been on the data safety monitoring board for EMD Serano, is a stockowner of Guardian, Shoreline, and RAPT Therapeutics, and has received travel reimbursement from RAPT Therapeutics. I. Ales Diaz, M. Ochoa-de-Olza, and S. Ponce Aix have nothing to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Trial inclusion and exclusion criteria.
Additional file 2.
Phase Ib baseline patient demographics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nadal, E., Saleh, M., Aix, S.P. et al. A phase Ib/II study of galunisertib in combination with nivolumab in solid tumors and non-small cell lung cancer. BMC Cancer 23, 708 (2023). https://doi.org/10.1186/s12885-023-11153-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-11153-1