
Abstract
Background
Dihydropyrimidine dehydrogenase (DPD) is a key enzyme in the metabolism of fluoropyrimidines. Variations in the encoding DPYD gene are associated with severe fluoropyrimidine toxicity and up-front dose reductions are recommended. We conducted a retrospective study to evaluate the impact of implementing DPYD variant testing for patients with gastrointestinal cancers in routine clinical practice in a high volume cancer centre in London, United Kingdom.
Methods
Patients receiving fluoropyrimidine chemotherapy for gastrointestinal cancer prior to, and following the implementation of DPYD testing were identified retrospectively. After November 2018, patients were tested for DPYD variants c.1905+1G>A (DPYD*2A), c.2846A>T (DPYD rs67376798), c.1679T>G (DPYD*13), c.1236G>A (DPYD rs56038477), c.1601G>A (DPYD*4) prior to commencing fluoropyrimidines alone or in combination with other cytotoxics and/or radiotherapy. Patients with a DPYD heterozygous variant received an initial dose reduction of 25–50%. Toxicity by CTCAE v4.03 criteria was compared between DPYD heterozygous variant and wild type carriers.
Results
Between 1st December 2018 and 31st July 2019, 370 patients who were fluoropyrimidine naïve underwent a DPYD genotyping test prior to receiving a capecitabine (n = 236, 63.8%) or 5FU (n = 134, 36.2%) containing chemotherapy regimen. 33 patients (8.8%) were heterozygous DPYD variant carriers and 337 (91.2%) were wild type. The most prevalent variants were c.1601G > A (n = 16) and c.1236G > A (n = 9). Mean relative dose intensity for the first dose was 54.2% (range 37.5–75%) for DPYD heterozygous carriers and 93.2% (42.9–100%) for DPYD wild type carriers. Overall grade 3 or worse toxicity was similar in DPYD variant carriers (4/33, 12.1%) as compared to wild-type carriers (89/337, 25.7%; P = 0.0924).
Conclusions
Our study demonstrates successful routine DPYD mutation testing prior to the initiation of fluoropyrimidine chemotherapy with high uptake. In patients with DPYD heterozygous variants with pre-emptive dose reductions, high incidence of severe toxicity was not observed. Our data supports routine DPYD genotype testing prior to commencement of fluoropyrimidine chemotherapy.
Similar content being viewed by others


Introduction
Fluoropyrimidines are integral chemotherapy drugs in the treatment of gastrointestinal cancers [1, 2]. In European oncology practice, the fluoropyrimidines 5-fluorouracil (5FU) and the orally bioavailable capecitabine are amongst the most commonly drugs prescribed as systemic therapy for tumours arising from the lower gastrointestinal tract (colon, rectal, anal canal) and upper digestive tracts (oesophagogastric, hepatopancreatic biliary). Fluoropyrimidines are also commonly used in the management of head and neck [3] and breast cancers [4].
Severe fluoropyrimidine chemotherapy toxicity occurs in approximately 30% of recipients [5]. Toxicity is characterised by myelosuppression, gastrointestinal toxicity (including diarrhoea and mucositis), hand and foot syndrome and cardiac toxicity [6,7,8]. Mortality from severe fluoropyrimidine toxicity occurs in 0.1–0.5% of patients [7, 9].
Dihydropyrimidine dehydrogenase (DPD) is a critical protein in the enzymatic degradation of fluoropyrimidines. As the rate limiting step in fluoropyrimidine clearance, 5FU is converted to 5-dihydrofluorouracil by DPD in the liver and responsible for 80–85% of the catabolism of fluoropyrimidines to inactive metabolites [10]. Genetic variants in the corresponding encoding DPYD gene occur in approximately 8% of patients of Caucasian ethnicity. The variants DPYD*2A (c.1905+1G>A), c.2846A>T, DPYD*13 (c.1679T>G) and c.1236G>A are associated with attenuated enzymatic activity and more frequent fluoropyrimidine related adverse events [11, 12].
Following the pivotal study by Henricks et al. [13], which demonstrated pre-emptive fluoropyrimidine dose reductions based upon DPYD variants reduced severe toxicity, DPYD variant testing and pre-emptive fluoropyrimidine chemotherapy dose reduction was implemented by the Gastrointestinal Unit at the Royal Marsden Hospital in November 2018. In this study, we conducted an audit to assess the implementation of a DPYD variant guided dosing and its effect on severe toxicity.
Methods
To identify patients receiving fluoropyrimidine chemotherapy, we interrogated the Royal Marsden Hospital pharmacy database for all prescriptions containing fluoropyrimidines (5FU and capecitabine) either in combination with other chemotherapy drugs and/or radiotherapy for gastrointestinal cancers after (1st December 2018 to 30th June 2019) DPYD testing implementation. Patients who had previously received systemic fluoropyrimidines were excluded from the study. To capture patients with homozygous DPYD variants, who may have not received fluoropyrimidines, we also searched for patients receiving raltitrexed, a non-fluoropyrimidine chemotherapeutic.
DPYD sequence variants were analysed in DNA extracted from EDTA whole blood. Common sequence variants DPYD c.1905+1G>A, c.2846A>T, c.1679T>G, c.1236G>A and c.1601G>A were genotyped by TaqMan assay (Applied Biosystems) using an AriaMx Real-Time PCR instrument (Agilent).
Treating physicians were provided with pathology reports which recommended initial dose reductions of 50% for heterozygous DPYD*2A or c.1679T>G variants; 25% for c.1236G>A and c.2846A>T; or 20% with a c.1601G>A variant. Patients with a DPYD heterozygous variant received an initial dose at the discretion of the treating physician. If initial doses of fluoropyrimidines were tolerated, doses of fluoropyrimdines were escalated for subsequent cycles. DPYD homozygous variant carriers did not receive fluoropyrimidines. DPYD wild-type carriers were dosed according to standard of care. Data on patient demographics, tumour, treatment and toxicity by CTCAE v4.03 criteria was collected retrospectively from electronic medical records, discharge summaries and chemotherapy charts.
The primary aim of the study was to compare the frequency of severe toxicity (grade ≥ 3) between DPYD wildtype and variant carriers with genotype based dosing. Other outcomes of interest were compliance with routine testing, turnaround time of DPYD testing, frequency of hospital admissions, treatment cessations and deaths due to fluoropyrimidine toxicity.
Comparisons of outcomes between groups were analysed using risk ratios and the Exact Fisher test. All P values were two-sided and a P value < 0.05 was considered statistically significant. Statistical analysis was performed on R-studio version 0.99.447.
Patient and public involvement
Patients and the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Results
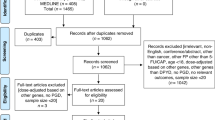
To compare the frequency of severe toxicities, we identified patients commencing fluoropyrimidine chemotherapy following implementation of routine DPYD testing in November 2018. Between 1st December 2018 and 31st July 2019, we identified 542 patients commencing fluoropyrimidine chemotherapy, of whom 150 patients were not receiving fluoropyrimidines for the first time. The remaining 392 patients were naïve to fluoropyrimidine chemotherapy. The DPYD guided cohort was comprised of the 370 patients (94.6%) who underwent, DPYD variant testing prior to receiving fluoropyrimidine containing chemotherapy. (Fig. 1).

CONSORT diagram Abbreviations: FP – fluoropyrimidine, DPYD – dihydropyrimidine dehydrogenase
Of the patients in the analysis (n = 370), the median age was 64 years (range 30–90). The majority were male (64.3%) and of white ethnicity (274, 74.1%). The predominant tumour type was colorectal cancer (n = 209, 56.5%), followed by oesophagogastric (n = 93, 25.1%) and hepatopancreatic biliary (n = 52, 14.1%). The majority of patients had an ECOG performance status of 0 (n = 100, 27%) or 1 (n = 254, 68.6%). The characteristics amongst the DPYD variant and wildtype cohorts were similar. (Table 1).
DPYD testing
For DPYD testing, the median time from blood draw to result was 6 days (IQR 5–7, range 0–18). Twenty-two DPYD tests were missed, with the majority of the missed tests (17/22, 77.2%) occurring with the first 2 months of testing implementation.
Amongst the 370 patients who underwent DPYD genotyping, 36 variants were detected amongst 33 patients (8.9%). The most common variants were c.1601G>A (n = 16, 4.3%), followed by c.1236G>A (n = 11, 3.0%), c.1905+ 1G > A (n = 4, 1.1%), c.2846A>T (n = 4, 1.1%) and c.1679T >G (n = 1, 0.3%). Thirty patients had a single heterozygous variant and 3 patients had a compound heterozygous variant. All compound heterozygous variants were associated with the c.1601G>A variant which co-occurred in two patients with c.1236G > A, and one patient with c.1905+1G>A. Concurrently, we identified 18 patients who received raltitrexed following DPYD genotyping implementation, of which none were carriers of homozygous DPYD variants.
Treatment
The majority of the patients were receiving fluoropyrimidine chemotherapy treatment with curative intent (n = 207, 55.9%) or as first line therapy (n = 359, 97%). Fluoropyrimidines were most commonly administered as a part of a chemotherapy doublet regimen (n = 219, 59.2%), single agent therapy (n = 92, 24.2%) or triplet therapy (n = 59, 15.9%). In 64 patients (17.3%), fluoropyrimidines were administered in combination with radiotherapy. Capecitabine was the most frequently prescribed fluoropyrimidine (n = 236, 63.8%), with the remainder of the patients receiving 5FU (n = 134, 36.2%). The proportions of characteristics were similar between the DPYD variant and wildtype cohorts. The relative dose intensities of the initial cycle of fluoropyrimidine was 54.2% (range 37.5–75%) for DPYD variant carriers, and 93.2% (42.9–100) for DPYD wildtype carriers. The two patients with compound heterozygous variants c.1236G>A/c.1601G>A commenced fluoropyrimidines at 50%. One patient with the compound heterozygous variant c.1905+1G>A/c.1601G>A was treated with an initial dose intensity of 41%. (Table 2).
DPYD wildtype vs DPYD variants
To understand the impact of pharmacogenomic guided dosing on DPYD variant carriers, we compared the toxicities of wildtype and variant carriers. In total, 4 patients (12.1%) in the DPYD variant cohort experienced grade ≥ 3 toxicity with 2 patients experiencing gastrointestinal toxicity and 1 patient with severe haematological toxicity. By comparison, 89 patients in the wildtype group experienced grade ≥ 3 toxicity with haematological (46, 13.6%) and gastrointestinal (29, 8.6%) the most frequent. Eleven patients, all in the wildtype cohort experienced cardiac toxicity of any grade. There were no statistically significant differences in the frequency of grade ≥ 3 adverse events between DPYD variant and wildtype carriers. (Table 3) In keeping with the intended dosing strategy, 10 patients (30.3%) in the DPYD variant cohort had a dose escalation after the first cycle of fluoropyrimidines whereas only 7 patients (2.1%) of the wildtype cohort received a dose escalation (P < 0.00001). Dose reductions were more common within the wildtype cohort compared with the variant cohort (18.3% vs 3.0%, P = 0.0261). The frequency of early treatment discontinuation (within the first two cycles of therapy) was similar in the wildtype and variant cohorts (3.3% vs 6.1%, P = 0.3245). Fluoropyrimidine dose reductions within the first two cycles of chemotherapy was also similar between both groups (6.1% vs 10.7%, P = 0.2282).
The clinical characteristics of the 4 carriers of DPYD variants experiencing severe fluoropyrimidine toxicity are detailed in Table 4. Three of these patients were receiving FOLFIRINOX (5FU, oxaliplatin, irinotecan) triplet chemotherapy which is frequency associated with severe toxicity [14]. In two patients, grade ≥ 3 toxicity occurred after 8 cycles of treatment which would be consistent with toxicity from non-fluoropyrimidine agents. Amongst DPYD wildtype carriers, grade ≥ 3 toxicity occurred in 10 patients (12.0%) receiving single agent chemotherapy and was significant higher in patients receiving doublet chemotherapy (53/198, 26.8%; P = 0.0074) and receiving triplet chemotherapy (26/56, 46.4%; P < 0.0001).
Discussion
Following the pivotal prospective study by Henricks et al. [13], we implemented routine DPYD pharmacogenomic guided dosing for patients commencing fluoropyrimidines for gastrointestinal cancers. Since initiating DPYD testing at the Royal Marsden Hospital, nationwide guidelines have been implemented with widely available DPYD genotype testing available through the NHS England Genomic Test Directory.
Our study demonstrates routine DPYD variant testing prior to the initiation of fluoropyrimidines can be successfully integrated into clinical practice. Compliance rates of DPYD testing was > 90% and the median laboratory turnaround time was 6 days. Patients were often tested for DPYD variants at the time of their initial medical oncology clinic appointment. Given the lead times for other investigations such as diagnostic imaging and chemotherapy day-unit scheduling, this turnaround time was not thought to cause a delay in the commencement of the chemotherapy. As the rollout of DPYD variant testing continues through the NHS England Genomic Test Directory, turnaround times are likely to improve. With the adoption of in-house testing, the turnaround time at our institution is now within 48 h.
The overall prevalence of the four DPYD variants recommended for testing (DPYD*2A, c.1236G>A, c.2846A>T, 1679G>A) amongst the population (20/330 5.4%) was slightly lower, but consistent with previous data in Caucasian populations. The c.1236A > T (HapB3) is the most prevalent clinically significant variant (2.4%), followed by c.1601G>A (DPYD*4) (2.0%), c.1905+1G>A (DPYD*2A) (0.8%), c.2846A>T (0.4%) and c.1679T>G (DPYD*13) (0.06%) [12]. The frequency of these variants are lower in other ethnic groups and further research is required to optimise a pharmacogenomic dosing approach in these populations [12].
In our clinical practice, we predominantly used 50% dose reductions of fluoropyrimidines with the first cycle of chemotherapy. Whilst this is recommended for heterozygous DPYD*2A and c.1679T >G variant carriers, we also extended this to carriers of other variants. We based this practice on prospective data suggesting 25% dose reductions are inadequate to mitigate toxicity with the c.1236G>A and c.2846A>T variants [13].
The incidence of grade ≥ 3 toxicity were not statistically different between patients with DPYD variants and wildtype. Taking into account the propensity for fluoropyrimidine toxicity in DPYD variant carriers, our result does suggest some level of effectiveness of upfront dose reductions. The incidence of severe fluoropyrimidine toxicity amongst carriers of DPYD variants was (4/33, 12.1%) and was similar to other real world case series of pharmacogenomic guided fluoropyrimidine dosing [15,16,17].
Despite pre-emptive fluoropyrimidine dose reductions, four patients with DPYD variants experienced severe toxicity due to the use of fluoropyrimidine with other cytotoxic drugs. Three of these patients were receiving FOLFIRINOX chemotherapy which is associated with high rates of severe toxicity [14]. Severe toxicity occurred in 25.7% DPYD wild type patients with doublet and 46.4% receiving triplet combination therapy. Whilst this may provide an argument for the limitations of DPYD variant testing, it does underline the need to develop pre-emptively strategies to reduce toxicity with fluoropyrimidine combination regimens. DPD phenotypic testing (uracil), which was not performed in this study may have identified additional patients at risk of fluoropyrimidine toxicity [18].
The most important limitation of our study is its retrospective design and sample size from a single institution. Only 33 (8.9%) carriers of DPYD variants were identified which is insufficient to provide a definitive estimate of the incidence of severe toxicities. Indeed, it is possible that the DPYD wildtype carriers have a different DPYD variant which was not detected with the available assays. Severe toxicity is also further confounded by the use of combination regimens which could be independent of DPYD variant status. Due to the low number of patients with DPYD variants, we could not perform meaningful analyses of populations at higher risk of toxicity including recipients of concurrent radiotherapy [19], female sex [20], age [21] and adjuvant/metastatic intent [22]. We did not record creatinine clearance which is a known risk factor particularly for capecitabine toxicity, however upfront reductions are routinely indicated in our practice in patients with lower creatinine clearance (30-50 mL/min). The reporting laboratory provided fluoropyrimidine dose reduction recommendations rather than the DPD activity score. [12] The use of the activity score provides user friendly dosing guidance particularly if multiple clinically significant DPYD variants are detected.
Following the implementation of pre-emptive DPYD variant testing, the routine testing of the c.1601G>A variant is no longer recommended. Whilst one case series has reported the c.1601G > A variant as a clinically significant predictor of fluoropyrimidine toxicity [23], this was not proven in a meta-analysis [11]. Furthermore, biochemical analysis suggests this variant does not result in loss of enzymatic activity [24].
In our study, we did not record the efficacy outcomes of the treatments. Due to the sample size and heterogeneity of the cohorts, it would have been insufficiently powered to demonstrate a conclusive result. A case–control study suggested a pre-emptive dose reduction strategy results in similar outcomes to patients with DPYD wildtype [25]. Furthermore, pharmacokinetic studies suggest AUC drug exposure with DPYD guided dosing is similar amongst variant and wildtype patients [13]. Whilst further study is necessary, the strategy of dose escalation based upon tolerance, was successful in ten patients (30.3%), and mitigates these efficacy concerns.
In conclusion, our study demonstrates successful implementation of routine DPYD variant testing amongst fluoropyrimidine naïve patients with gastrointestinal cancers. Severe toxicity amongst DPYD variant carriers was not observed and had comparable rates of toxicity to DPYD variant carriers. Our data supports routine testing and pre-emptive dosing strategies which is now standardly done within the NHS.
Availability of data and materials
Data from this study can be made available to other researchers upon reasonable request and subject to data transfer agreements. Requests should be directed to NS.
References
Yoshino T, Arnold D, Taniguchi H, Pentheroudakis G, Yamazaki K, Xu RH, Kim TW, Ismail F, Tan IB, Yeh KH, et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO-ESMO initiative endorsed by CSCO, KACO, MOS SSO and TOS. Ann Oncol. 2018;29(1):44–70.
NCCN: Colon Cancer v2.2021. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed 3 Dec 2021.
Machiels JP, René Leemans C, Golusinski W, Grau C, Licitra L, Gregoire V. Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(11):1462–75.
Gennari A, André F, Barrios CH, Cortés J, de Azambuja E, DeMichele A, Dent R, Fenlon D, Gligorov J, Hurvitz SA, et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer<sup>☆</sup>. Ann Oncol. 2021;32(12):1475–95.
Froehlich TK, Amstutz U, Aebi S, Joerger M, Largiadèr CR. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int J Cancer. 2015;136(3):730–9.
Chionh F, Lau D, Yeung Y, Price T, Tebbutt N. Oral versus intravenous fluoropyrimidines for colorectal cancer. Cochrane Database Syst Rev. 2017;7(7):Cd008398.
Lévy E, Piedbois P, Buyse M, Pignon JP, Rougier P, Ryan L, Hansen R, Zee B, Weinerman B, Pater J, et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol. 1998;16(11):3537–41.
Tebbutt NC, Wilson K, Gebski VJ, Cummins MM, Zannino D, van Hazel GA, Robinson B, Broad A, Ganju V, Ackland SP, et al. Capecitabine, bevacizumab, and mitomycin in first-line treatment of metastatic colorectal cancer: results of the Australasian Gastrointestinal Trials Group Randomized Phase III MAX Study. J Clin Oncol. 2010;28(19):3191–8.
Sharma BB, Rai K, Blunt H, Zhao W, Tosteson TD, Brooks GA. Pathogenic DPYD Variants and Treatment-Related Mortality in Patients Receiving Fluoropyrimidine Chemotherapy: A Systematic Review and Meta-Analysis. Oncologist. 2021;26(12):1008–16.
Miura K, Kinouchi M, Ishida K, Fujibuchi W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, et al. 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers (Basel). 2010;2(3):1717–30.
Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U, Largiadèr CR, Jennings BA, Marinaki AM, Sanderson JD, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16(16):1639–50.
Amstutz U, Henricks LM, Offer SM, Barbarino J, Schellens JHM, Swen JJ, Klein TE, McLeod HL, Caudle KE, Diasio RB, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther. 2018;103(2):210–6.
Henricks LM, Lunenburg C, de Man FM, Meulendijks D, Frederix GWJ, Kienhuis E, Creemers GJ, Baars A, Dezentjé VO, Imholz ALT, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459–67.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med. 2011;364(19):1817–25.
Wigle TJ, Povitz BL, Medwid S, Teft WA, Legan RM, Lenehan J, Nevison S, Panuganty V, Keller D, Mailloux J, et al. Impact of pretreatment dihydropyrimidine dehydrogenase genotype-guided fluoropyrimidine dosing on chemotherapy associated adverse events. Clin Transl Sci. 2021;14(4):1338–48.
Jolivet C, Nassabein R, Soulières D, Weng X, Amireault C, Ayoub JP, Beauregard P, Blais N, Carrier C, Cloutier AS, et al. Implementing DPYD*2A Genotyping in Clinical Practice: The Quebec, Canada. Experience Oncologist. 2021;26(4):e597–602.
Wang L, Howlett S, Essapen S. Treating patients with dihydropyrimidine dehydrogenase (DPD) deficiency with fluoropyrimidine chemotherapy since the onset of routine prospective testing-The experience of a large oncology center in the United Kingdom. Semin Oncol. 2022;49(2):170–7.
Meulendijks D, Henricks LM, Jacobs BAW, Aliev A, Deenen MJ, de Vries N, Rosing H, van Werkhoven E, de Boer A, Beijnen JH, et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity. Br J Cancer. 2017;116(11):1415–24.
De Caluwé L, Van Nieuwenhove Y, Ceelen WP. Preoperative chemoradiation versus radiation alone for stage II and III resectable rectal cancer. Cochrane Database Syst Rev. 2013(2):Cd006041.
Sloan JA, Goldberg RM, Sargent DJ, Vargas-Chanes D, Nair S, Cha SS, Novotny PJ, Poon MA, O’Connell MJ, Loprinzi CL. Women experience greater toxicity with fluorouracil-based chemotherapy for colorectal cancer. J Clin Oncol. 2002;20(6):1491–8.
Knikman JE, Gelderblom H, Beijnen JH, Cats A, Guchelaar H-J, Henricks LM. Individualized Dosing of Fluoropyrimidine-Based Chemotherapy to Prevent Severe Fluoropyrimidine-Related Toxicity: What Are the Options? Clin Pharmacol Ther. 2021;109(3):591–604.
Baird R, Biondo A, Chhaya V, McLachlan J, Karpathakis A, Rahman S, Barbachano Y, Cunningham D, Chau I. Toxicity associated with capecitabine plus oxaliplatin in colorectal cancer before and after an institutional policy of capecitabine dose reduction. Br J Cancer. 2011;104(1):43–50.
Loganayagam A, Arenas Hernandez M, Corrigan A, Fairbanks L, Lewis CM, Harper P, Maisey N, Ross P, Sanderson JD, Marinaki AM. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer. 2013;108(12):2505–15.
Offer SM, Fossum CC, Wegner NJ, Stuflesser AJ, Butterfield GL, Diasio RB. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74(9):2545–54.
Henricks LM, van Merendonk LN, Meulendijks D, Deenen MJ, Beijnen JH, de Boer A, Cats A, Schellens JHM. Effectiveness and safety of reduced-dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: A matched pair analysis. Int J Cancer. 2019;144(9):2347–54.
Acknowledgements
We would like to acknowledge the generosity of the patients who participated in this study.
We would like to acknowledge the National Institute for Health Research (NIHR) Biomedical Research Centre at The Royal Marsden NHS Foundation Trust and the Institute of Cancer Research, London.
The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Funding
This research received no specific grant from any funding agency in public, commercial or no-for-profit sectors.
Author information
Authors and Affiliations
Contributions
DL, C Fong, FA and NS conceptualised and designed the study. DL, C Fong, FA, LC, HK, RGE, MBR, SL, AMD collected the data. DL, CF and NS carried out the statistical analyses. DL drafted the manuscript. All authors provided data interpretation, reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. NS accepts full responsibility for the finished work and the conduct of the study, had access to the data, and the final decision to publish. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Committee for Clinical Research at the Royal Marsden Hospital and informed consent of participant was waived due to retrospective nature of study. The study was conducted in accordance with the principles laid down by the World Medical Assembly in the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
DL is the recipient of the Australasian Gastro-Intestinal Trials Group/Merck Clinical Research Fellowship. SR has served in a consulting role or on the advisory boards for Bayer, Boehringer Ingelheim, Hookipa Biotech, Incyte and Merck Serono.IC has served in a consulting role or on the advisory boards for Eli Lilly, Bristol Meyers Squibb, MSD, Bayer, Roche, Merck Serono, Five Prime Therapeutics, Astra Zeneca, OncXerna, Pierre Fabre, Boehringer Ingelheim, Incyte, Astellas, GSK, Sotio, Eisai and Daiichi Sankyo; received research funding from Eli Lilly, Janssen-Cilag; and honoraria from Eli Lilly, Eisai and Servier.DC has served in a consulting role or on the advisory boards for OVIBIO; received institutional research funding from 4SC, Bayer, Celgene, Clovis Oncology, Leap Oncology, Eli Lilly, MedImmune, Roche; and has ownership interests in OVIBIO.NS has served in a consulting role or on the advisory boards for AstraZeneca, MSD, Pfizer, Servier; received institutional research funding from AstraZeneca, BMS, Pfizer/ EMD Serono; honoraria from Amgen, GlaxoSmithKline, Lilly, Merck Serono, MSD Oncology, Pierre Fabre, Servier; and travel accommodation expenses from AstraZeneca, BMS, Lilly, Merck, MSD Oncology and Roche. C Fong, FA, LC, HK, RGE, MBR, SL, AMD, MAH, MH, C Fribbens and DW have no disclosures.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lau, D.K., Fong, C., Arouri, F. et al. Impact of pharmacogenomic DPYD variant guided dosing on toxicity in patients receiving fluoropyrimidines for gastrointestinal cancers in a high-volume tertiary centre. BMC Cancer 23, 380 (2023). https://doi.org/10.1186/s12885-023-10857-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-10857-8