
Abstract
Background
Previous large observational cohort studies showed higher blood pressure (BP) positively associated with cancer. We used Mendelian randomization (MR) to obtain less confounded estimates of BP on total and site-specific cancers.
Methods
We applied replicated genetic instruments for systolic and diastolic BP to summary genetic associations with total cancer (37387 cases, 367856 non-cases) from the UK Biobank, and 17 site-specific cancers (663–17881 cases) from a meta-analysis of the UK Biobank and the Kaiser Permanente Genetic Epidemiology Research on Adult Health and Aging. We used inverse-variance weighting with multiplicative random effects as the main analysis, and sensitivity analyses including the weighted median, MR-Egger and multivariable MR adjusted for body mass index and for smoking. For validation, we included breast (Breast Cancer Association Consortium: 133384 cases, 113789 non-cases), prostate (Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome Consortium: 79194 cases, 61112 non-cases) and lung (International Lung and Cancer Consortium: 10246 cases, 38295 non-cases) cancer from large consortia. We used asthma as a negative control outcome.
Results
Systolic and diastolic BP were unrelated to total cancer (OR 0.98 per standard deviation higher [95% confidence interval (CI) 0.89, 1.07] and OR 1.00 [95% CI 0.92, 1.08]) and to site-specific cancers after accounting for multiple testing, with consistent findings from consortia. BP was nominally associated with melanoma and possibly kidney cancer, and as expected, not associated with asthma. Sensitivity analyses using other MR methods gave similar results.
Conclusions
In contrast to previous observational evidence, BP does not appear to be a risk factor for cancer, although an effect on melanoma and kidney cancer cannot be excluded. Other targets for cancer prevention might be more relevant.
Similar content being viewed by others


Background
Elevated blood pressure (BP), or hypertension, reduces population health globally [1], with 31.1% of the world’s adult population estimated to be hypertensive in 2010 [2], and 10.4 million deaths worldwide attributed to high systolic BP in 2016 [3]. In addition to the well-established relation of BP with cardiovascular disease (CVD) [4], hypertension has been linked with higher risk of cancer observationally [5,6,7], but the evidence is inconsistent with the possible exception of kidney cancer [8]. Secondary analyses of randomized controlled trials (RCTs) of antihypertensive drugs found little association with cancer [9], but RCTs typically have follow-up times too short to detect effects on cancer risk. Although the underlying mechanisms linking hypertension to cancer are still unclear, it has been suggested that increased cell turnover and telomere shortening could play a role [10]. In addition, dysregulated immune function is implicated in the pathogenesis of both hypertension and cancer [11, 12], and BP is positively associated with white blood cell count [13]. Nevertheless, confounding by social and environmental factors could give rise to the observed associations [14]. Mendelian randomization (MR), by using genetic variants randomly allocated at conception as instrumental variables, is less susceptible to confounding than conventional observational studies [15]. In this MR study using two-sample methods, we assessed the effects of systolic and diastolic BP on total cancer as well as on 17 common site-specific cancers, by applying replicated genetic instruments for BP to large population-based cohorts. For validation, we included large genetic consortia for breast, prostate and lung cancer. We also used multivariable MR [16, 17] to mitigate potential pleiotropic effects via obesity and smoking.
Methods
Genetic instruments for blood pressure
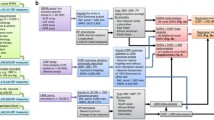
We extracted strong (P < 5x10-8), independent (r2 < 0.001) and externally replicated single nucleotide polymorphisms (SNPs) predicting BP from a meta-analysis of genome-wide association studies (GWAS) for BP traits totaling 757,601 participants of European ancestry (mean age 56.0 years, 54.7% women) [18], consisting of 458,577 individuals from the UK Biobank excluding pregnant women (n=372) and individuals who had withdrawn consent (n=36) [19], and 299,024 individuals from an enlarged dataset of the International Consortium for Blood Pressure (ICBP) with 77 cohorts [20]. Independent replication included 220,520 individuals from the Million Veteran Program [21] and 28,742 individuals from the Estonian Biobank of the Estonian Genome Center University of Tartu [22]. Participants on BP lowering medication had their BP values adjusted by adding 15 and 10 mm Hg to systolic and diastolic BP, respectively [23]. The UK Biobank analysis used a linear mixed model [24], adjusted for age, age2, sex and body mass index (BMI), with genomic control applied at the study level to correct for inflation due to population stratification and cryptic relatedness [25], followed by fixed-effect meta-analysis with the ICBP summary statistics which also adjusted for the same covariates. The pooled mean (standard deviation (SD)) systolic and diastolic BP were 138.4 (20.1) and 82.8 (11.2) mm Hg, respectively. The BP GWAS adjusted for BMI, which could bias the estimates of genetic variants on BP if genetic variants or environmental factors driving both BMI and BP exist [26], and potentially the MR estimates and/or instrument selection. We repeated the analysis for total cancer using genetic predictors from the UK Biobank, which did not adjust for BMI.
Genetic associations with total and site-specific cancers
Genetic associations with total cancer (phenocode:195) were obtained from a pan-ancestry GWAS of the UK Biobank [27], with lifetime cancer occurrence ascertained from linked medical records (hospital inpatient data and death registry) including both prevalent and incident cases [28]. MR studies evaluate lifelong effects of an exposure, and so necessitate the inclusion of lifelong cases in consideration of potential selection bias [29]. Of the 441,331 participants included, genetic associations were provided for the 420,531 (95.3%) individuals of European ancestry to minimize confounding by population stratification. Non-cases were individuals without a diagnosis of primary or secondary cancer, nor a history of radio- or chemotherapy. The analyses used the Scalable and Accurate Implementation of Generalized mixed model, which accounts for sample relatedness and extreme case-control ratio [30], and adjusted for age, sex, age*sex, age2, age2*sex and the first 10 principal components (PCs).
Genetic associations with site-specific cancers were obtained from the largest available pan-cancer GWAS [31], which provides summary genetic associations with 17 cancers for 475,312 individuals of European ancestry from the UK Biobank and the Kaiser Permanente Genetic Epidemiology Research on Adult Health and Aging (GERA) [32, 33]. Lifetime cancer occurrence was ascertained from linked medical records with the latest diagnosis in August 2015 in the UK Biobank and June 2016 in GERA, which were converted into the third revision of International Classification of Diseases for Oncology (ICD-O-3) codes and classified according to organ site based on the U.S. National Cancer Institute Surveillance, Epidemiology, and End Results Program recode paradigm [34]. The median age at diagnosis was lowest for cervical cancer (37 and 38 years in the UK Biobank and GERA, respectively) and highest for pancreatic cancer (66 and 76 years). Individuals with multiple diagnoses were only recorded for their first cancer. Non-cases were cancer-free individuals, i.e., those who did not have any cancer diagnosis, self-reported history of cancer or cancer as a cause of death. For sex-specific cancer (breast, cervix, endometrium, ovary, prostate and testis), same-sex non-cases were used. Summary genetic associations are available for bladder, breast, cervix, colon, esophagus/stomach, kidney, lung, lymphocytic leukemia, melanoma, non-Hodgkin lymphoma, oral cavity/pharyngeal, ovary, pancreas, prostate, rectum and thyroid. The analyses were conducted separately for each cohort using logistic regression, adjusted for age, sex, the first 10 PCs, genotyping array (UK Biobank only) and reagent kit for genotyping (GERA only), followed by meta-analysis. Standard error (SE) of the SNP-outcome association were estimated from the p-value [35], as it was not provided.
For validation of potentially small effects, we additionally included large genetic consortia of leading cancers [36], i.e., breast (133384 cases and 113789 non-cases) [37], prostate (79194 cases and 61112 non-cases) [38] and lung (10246 cases and 38295 non-cases) [39], which have larger number of cases and do not overlap with the UK Biobank or GERA.
Estimates were aligned on the same effect allele for BP and cancer. Effect allele frequency (EAF) was not provided for pan-cancer, so we used the UK Biobank EAF which constituted 86% of the participants. Palindromic SNPs with ambiguous EAF, i.e. >0.42 and <0.58, and SNPs instrumenting BP but not available for an outcome, were replaced by proxies (r2 ≥ 0.8) identified using LDlink [40], wherever available.
Genetic associations with asthma
Genetic associations with asthma (31169 cases and 379656 non-cases) used as a negative control outcome were obtained from the UK Biobank (phenocode: 495), given BP is not known to cause asthma [41], but both share similar confounders [42].
Statistical analysis
Instrument strength was assessed using the F-statistic [43], approximated by the squared SNP-phenotype association divided by its variance. An F-statistic < 10 suggests potentially weak instrument. We also estimated the I2 to assess heterogeneity of instrument strength, an I2 < 90% suggests violation of the no measurement error (NOME) assumption and possibly invalid estimates [43]. An I2 > 97% suggests minimal bias of the MR estimates by confounding of exposure on outcome in overlapping samples [44], as here. The proportion of phenotypic variance (r2) explained by the genetic instruments was calculated as beta2*2*MAF*(1-MAF), where beta is the SNP-phenotype association standardized to the phenotypic variance and MAF is the minor allele frequency of the SNP [45]. Power calculations were based on the approximation that the sample size for an MR study is the sample size for exposure on outcome divided by the r2 for genetic instruments on exposure [46], using an online tool [47].
We used the inverse-variance weighted (IVW) meta-analysis, with multiplicative random effects, which assumes balanced pleiotropy [48], of the SNP-specific Wald estimates, i.e., the SNP-outcome association divided by the SNP-exposure association, as the main analysis. We also conducted sensitivity analyses using the weighted median [49] and MR-Egger [50]. The weighted median assumes 50% of the weight is from valid SNPs. MR-Egger is robust to genetically invalid instruments given the instrument strength Independent of direct effect (InSIDE), i.e., the instruments do not confound exposure on outcome, and the NOME assumption is satisfied. A zero MR-Egger intercept indicates evidence of lack of such genetic pleiotropy. Some of the genetic instruments for BP was previously shown to be associated with confounders of BP and cancer, mostly for anthropometrics and a few for lifestyle [18], so we used multivariable MR to estimate the effects of BP on cancer independent of BMI or ever-smoking using IVW or MR-Egger if the intercept was non-zero. We obtained genetic associations with BMI and ever-smoking from Yengo et al. [51], and the Social Science Genetic Association Consortium [52], respectively. We dropped correlated predictors of BP and BMI or ever-smoking. We estimated the Sanderson-Windmeijer multivariate F-statistic and modified Q statistic to assess conditional instrument strength and heterogeneity, taking into account the phenotypic correlation [53], using estimates from the UK Biobank [54].
All analyses were performed using R (version 4.0.1, The R Foundation for Statistical Computing Platform, Vienna, Austria). We used the R packages “TwoSampleMR”, “MedelianRandomization” and “MVMR”. Given the number of cancer outcomes considered, a two-sided p-value below the Bonferroni-corrected significance threshold 0.0014 (0.05/2 BP traits*18 cancer outcomes) was used.
Results
There were 272 and 267 strong, independent and replicated SNPs predicting systolic (Supplementary Table S1) and diastolic (Supplementary Table S2) BP with a mean (range) F-statistic of 83.2 (29.3 – 612.4) and 90.7 (30.0 – 818.1), and I2 of 92.5 and 93.8%, respectively (Table 1). These SNPs explained approximately 2.59 and 2.96% of the variance of systolic and diastolic BP, respectively. At 5% alpha, this study has 80% power to detect an odds ratio (OR) of about 1.09 for total cancer, and from 1.13 for breast to 1.60 for thyroid cancer per SD of BP (Supplementary Table S3). We obtained 539 and 92 strong (P < 5x10-8) and independent (r2 < 0.001) SNPs predicting BMI and ever-smoking, respectively. Both BMI (Supplementary Fig. S1) and ever-smoking (Supplementary Fig. S2) were positively associated with total cancer. The Sanderson-Windmeijer multivariate F-statistics were at least 33.8 for systolic and 36.9 for diastolic BP when adjusted for BMI, and at least 75.3 for systolic and 80.8 for diastolic BP when adjusted for ever-smoking.
Figure 1 shows the associations of systolic and diastolic BP (per 1-SD increment) with total cancer. Overall, systolic (OR 0.98 [95% confidence interval (CI) 0.89, 1.07] and diastolic BP (OR 1.00 [95% CI 0.92, 1.08]) were not associated with total cancer. Repeating the analysis using genetic instruments unadjusted for BMI, or sensitivity analysis using the weighted median, MR-Egger and multivariable MR gave similar results (Supplementary Tables S4 and S5).

Mendelian randomization (MR) estimates of systolic and diastolic BP on total cancer. BMI, body mass index; IVW, inverse variance weighting
Systolic and diastolic BP were not significantly associated with any of the 17 site-specific cancers in the meta-analysis of the UK Biobank and GERA. Some associations at nominal significance were observed for kidney cancer and melanoma (Fig. 2). Similarly, no significant associations were observed for breast, prostate or lung cancer (Fig. 3) in the consortia, although systolic BP was nominally associated with lung cancer. Using other MR methods gave similar results for site-specific cancers (Supplementary Tables S4-6). Systolic and diastolic BP were not associated with asthma (Supplementary Fig. S3).

Inverse-variance weighted Mendelian randomization estimates of systolic and diastolic BP on 17 site-specific cancer

Inverse-variance weighted Mendelian randomization estimates of systolic and diastolic BP on breast, prostate and lung cancers in genetic consortia
Discussion
Consistent with secondary analyses of RCTs [9], but less consistent with observational studies [5,6,7,8], this MR study found little evidence of BP increasing risk of cancer. However, BP was nominally positively associated with kidney cancer [55, 56], and possibly melanoma [57]. As expected, BP was not associated with asthma.
This is the first MR study that has comprehensively evaluated the effect of BP on cancer. The main strength of the study is the MR design which minimizes confounding [58]. Long-term exposure to common risk factors for cancer [14], such as socio-economic position and all it entails, including smoking [59], alcohol consumption [60], diets promoting obesity [61], and air pollution [62] are known to elevate BP. So, previous observational findings showing higher BP positively associated with risk of total and some site-specific cancers [5,6,7,8], might be due to confounding by these factors. Sustained hypertension leads to compensatory vascular hypertrophy involving Angiotensin II mediated by various growth factors [63]. Angiotensin II receptors are found in high density in the kidney responsible for BP regulation [64]. A previous MR showed diastolic BP, but not systolic BP, was associated with higher risk of kidney cancer [56]. We did not replicate these findings at statistical significance, but only with directionally concordant results. Here, we used a different GWAS for kidney cancer, which had fewer cases, and adjusted for more covariates, such as age, to control for population structure. Further studies with more cases of kidney cancer would be helpful. Similarly, tumour growth following melanoma cell grafting was slower in Angiotensin II receptor deficient mice than in wild types [65]. Observationally, BP was positively associated melanoma [57], and further MR studies with larger samples are warranted. Although MR is less susceptible to confounding than traditional observation studies, it is not free from selection bias [66]. Specifically, given BP strongly reduces survival, some MR estimate may be attenuated by missing people who died before recruitment from genetically higher BP, from cancer or from a competing risk for cancer [67], such as CVD, particularly for cancers typically identified at older ages, including kidney cancer, prostate cancer and melanoma. As such, the estimate for total cancer could be a false negative. However, most cancer deaths typically occur at a younger age than deaths from other major causes [68], such as CVD, reducing this possibility.
Despite using a design less open to confounding than purely observational studies, and assessing associations independent of BMI as well as of smoking, our study has several limitations. First, the validity of MR rests on the three instrumental variable assumptions, i.e., the genetic instruments strongly predict the exposure, the genetic instruments are not associated with confounders of the exposure and outcome, and the genetic instruments are associated with the outcome only through affecting the exposure [69]. We used the largest available GWAS with external replication to obtain genetic instrument for BP, and sensitivity analysis to assess the robustness of our estimates, which were largely consistent. We also included a negative control outcome and did not find evidence of substantial pleiotropic effects. Second, the UK Biobank contributed information to the exposure and outcome GWAS, which may bias the MR estimates towards the observational association [70] particularly for weak instruments. However, weak instrument bias is inversely proportional to the F-statistics, which was only around 1%. Bias from confounding is unlikely to affect the analysis and would not explain the null findings [44]. Third, total cancer was based on incident and prevalent cases which might over-represent people living with treatable cancers. However, cancers have common underlying molecular hallmarks [71], whether BP might affect these hallmarks is unclear. In addition to total cancer, we investigated the effects of BP on 17 site-specific cancers. Although we found no association of BP with total cancer, we cannot rule out the possibility that BP has some specific effects on some site-specific cancers, which we could not reliably test owing to the small number of cases for some cancers. Furthermore, the grouping of heterogenous cancer sites, such as esophagus and stomach, limited the interpretation of some of our estimates, but were included for completeness. We additionally included large genetic consortia for breast, prostate and lung cancer for validation. Notably, these GWAS did not adjust for age, and the cases were on average younger than the non-cases, which could confound the MR estimates likely away from the null. Fourth, the BP instruments were adjusted for BMI. Adjusting for an effect of the exposure does not necessarily create bias [72], correspondingly genetic estimates for BP have been shown to be similar with and without adjustment for BMI [73]. Using genetic instruments for BP without adjustment for BMI also made little difference to the estimate for total cancer. Fifth, the UK Biobank is self-selected and differs from its underlying population in several major health and socioeconomic characteristics [74]. However, risk factor-outcome associations are comparable in the UK biobank and other UK-based studies with less self-selection [75]. Sixth, the present study included only participants of European ancestry, which avoids genetic confounding due to population stratification but may limit external validity in other ethnic groups. However, BP is not thought to act differently by ethnicity [76].
Globally, BP has been falling, most notably in high sociodemographic index countries in Asia Pacific and the West [77]. However, cancer rates are still rising even after taking into account population aging [78]. Obesity prevalence has been rising in both children and adults [79], which may instead underlie some of the rising cancer incidence, as well as raising BP. From a population health perspective, our findings are largely consistent with the absence of hypertension as an intervention target for primary cancer prevention [80]. Although this may undermine the importance of hypertension as a risk factor for health, it is perhaps more important that the benefits of BP lowering be accurately mapped out for evidence-based health promotion [81].
Conclusions
In this MR study, BP does not appear to be a risk factor for total cancer contrary to previous observational evidence, although an effect on melanoma and kidney cancer cannot be excluded. Other targets for cancer prevention might be more relevant.
Availability of data and materials
We thank the participants and researchers for providing the publicly available summary data used in this study. Data on blood pressure were downloaded from GWAS Catalog (ebi.ac.uk/gwas/); data on total cancer were downloaded from the UK Biobank (pan.ukbb.broadinstitute.org/); data on site-specific cancers were downloaded from github.com/Wittelab/pancancer_pleiotropy; data on breast cancer were downloaded from bcac.ccge.medschl.cam.ac.uk; data on prostate cancer were downloaded from practical.icr.ac.uk; data on lung cancer were downloaded from gwas.mrcieu.ac.uk; data on BMI were downloaded from portals.broadinstitute.org/collaboration/giant; data on ever-smoking were downloaded from the Social Science Genetic Association Consortium (thessgac.org).
Abbreviations
- BMI:
-
Body mass index
- BP:
-
Blood pressure
- CI:
-
Confidence interval
- CVD:
-
Cardiovascular disease
- EAF:
-
Effect allele frequency
- GERA:
-
Kaiser Permanente Genetic Epidemiology Research on Adult Health and Aging
- GWAS:
-
Genome-wide association studies
- ICBP:
-
International Consortium for Blood Pressure
- ICD-O-3:
-
Third revision of International Classification of Diseases for Oncology
- IVW:
-
Inverse-variance weighted
- InSIDE:
-
Instrument strength Independent of direct effect
- MAF:
-
Minor allele frequency
- MR:
-
Mendelian randomization
- NOME:
-
No measurement error
- OR:
-
Odds ratio
- PC:
-
Principal component
- RCT:
-
Randomized controlled trial
- SD:
-
Standard deviation
- SE:
-
Standard error
- SNP:
-
Single nucleotide polymorphism
References
Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, et al. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. JAMA. 2017;317(2):165–82.
Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, et al. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. 2016;134(6):441–50.
Gakidou E, Afshin A, Abajobir AA, Abate KH, Abbafati C, Abbas KM, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1345–422.
Wright JT Jr, Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103–16.
Stocks T, Van Hemelrijck M, Manjer J, Bjørge T, Ulmer H, Hallmans G, et al. Blood pressure and risk of cancer incidence and mortality in the Metabolic Syndrome and Cancer Project. Hypertension. 2012;59(4):802–10.
Christakoudi S, Kakourou A, Markozannes G, Tzoulaki I, Weiderpass E, Brennan P, et al. Blood pressure and risk of cancer in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer. 2020;146(10):2680–93.
Drozd M, Pujades-Rodriguez M, Sun F, Franks KN, Lillie PJ, Witte KK, Kearney MT, Cubbon RM. Causes of Death in People With Cardiovascular Disease: A UK Biobank Cohort Study. J Am Heart Assoc. 2021;10(22):e023188.
Seretis A, Cividini S, Markozannes G, Tseretopoulou X, Lopez DS, Ntzani EE, et al. Association between blood pressure and risk of cancer development: a systematic review and meta-analysis of observational studies. Sci Rep. 2019;9(1):8565.
Bangalore S, Kumar S, Kjeldsen SE, Makani H, Grossman E, Wetterslev J, et al. Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol. 2011;12(1):65–82.
Hamet P. Cancer and hypertension: a potential for crosstalk? J Hypertens. 1997;15(12 Pt 2):1573–7.
Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40.
Palmer S, Albergante L, Blackburn CC, Newman TJ. Thymic involution and rising disease incidence with age. 2018;115(8):1883–8.
Siedlinski M, Jozefczuk E, Xu X, Teumer A, Evangelou E, Schnabel RB, et al. White blood cells and blood pressure. Circulation. 2020;141(16):1307–17.
Wild C, Weiderpass E, Stewart BJLIAfRoC: World cancer report: cancer research for cancer prevention. 2020.
Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol. 2003;32(1):1–22.
Burgess S, Thompson SG. Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–60.
Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol. 2018;48(3):713–27.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25.
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779.
Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet. 2016;48(10):1171–84.
Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, et al. Million veteran program: a mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–23.
Leitsalu L, Haller T, Esko T, Tammesoo M-L, Alavere H, Snieder H, et al. Cohort profile: Estonian biobank of the Estonian Genome Center, University of Tartu. Int J Epidemiol. 2014;44(4):1137–47.
Tobin MD, Sheehan NA, Scurrah KJ, Burton PR. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med. 2005;24(19):2911–35.
Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284–90.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–5.
Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL, Kraft P. Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am J Hum Genet. 2015;96(2):329–39.
Pan-UKB team: https://pan.ukbb.broadinstitute.org. In.; 2020.
Wu P, Gifford A, Meng X, Li X, Campbell H, Varley T, et al. Mapping ICD-10 and ICD-10-CM Codes to phecodes: workflow development and initial evaluation. JMIR Med Inform. 2019;7(4):e14325.
Hernán MA, Sauer BC, Hernández-Díaz S, Platt R, Shrier I. Specifying a target trial prevents immortal time bias and other self-inflicted injuries in observational analyses. J Clin Epidemiol. 2016;79:70–5.
Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50(9):1335–41.
Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun. 2020;11(1):4423.
Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics. 2015;200(4):1285–95.
Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, Connell S, et al. Genotyping informatics and quality control for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics. 2015;200(4):1051–60.
National Cancer Institute: Site Recode ICD-O-3/WHO 2008 definition.
Altman DG, Bland JM. How to obtain the confidence interval from a P value. BMJ. 2011;343:d2090.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Zhang H, Ahearn TU, Lecarpentier J, Barnes D, Beesley J, Qi G, et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet. 2020;52(6):572–81.
Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet. 2018;50(7):928–36.
Wang Y, McKay JD, Rafnar T, Wang Z, Timofeeva MN, Broderick P, et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat Genet. 2014;46(7):736–41.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–7.
Wan EYF, Fung WT, Schooling CM, Au Yeung SL, Kwok MK, Yu EYT, et al. Blood pressure and risk of cardiovascular disease in UK biobank: a mendelian randomization study. Hypertension. 2021;77(2):367–75.
Lipsitch M, Tchetgen Tchetgen E, Cohen T. Negative controls: a tool for detecting confounding and bias in observational studies. Epidemiology. 2010;21(3):383–8.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Minelli C, Del Greco MF, van der Plaat DA, Bowden J, Sheehan NA, Thompson J. The use of two-sample methods for Mendelian randomization analyses on single large datasets. Int J Epidemiol. 2021.
Guan W, Steffen BT, Lemaitre RN, Wu JHY, Tanaka T, Manichaikul A, et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet. 2014;7(3):321–31.
Freeman G, Cowling BJ, Schooling CM. Power and sample size calculations for Mendelian randomization studies using one genetic instrument. Int J Epidemiol. 2013;42(4):1157–63.
Brion M-JA, Shakhbazov K, Visscher PM. Calculating statistical power in mendelian randomization studies. Int J Epidemiol. 2012;42(5):1497–501.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data mendelian randomization. Stat Med. 2017;36(11):1783–802.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641–9.
Karlsson Linnér R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51(2):245–57.
Sanderson E, Spiller W, Bowden J. Testing and correcting for weak and pleiotropic instruments in two-sample multivariable mendelian randomisation. bioRxiv 2020.
Zheng J, Richardson TG, Millard LAC, Hemani G, Elsworth BL, Raistrick CA, et al. PhenoSpD: an integrated toolkit for phenotypic correlation estimation and multiple testing correction using GWAS summary statistics. Gigascience. 2018;7(8).
Chow WH, Gridley G, Fraumeni JF Jr, Järvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med. 2000;343(18):1305–11.
Johansson M, Carreras-Torres R, Scelo G, Purdue MP, Mariosa D, Muller DC, et al. The influence of obesity-related factors in the etiology of renal cell carcinoma-A mendelian randomization study. PLoS Med. 2019;16(1):e1002724.
Nagel G, Bjørge T, Stocks T, Manjer J, Hallmans G, Edlinger M, et al. Metabolic risk factors and skin cancer in the Metabolic Syndrome and Cancer Project (Me-Can). Br J Dermatol. 2012;167(1):59–67.
Lawlor DA, Smith GD, Ebrahim S. Socioeconomic position and hormone replacement therapy use: explaining the discrepancy in evidence from observational and randomized controlled trials. Am J Public Health. 2004;94(12):2149–54.
Oncken CA, White WB, Cooney JL, Van Kirk JR, Ahluwalia JS, Giacco S. Impact of smoking cessation on ambulatory blood pressure and heart rate in postmenopausal women*. Am J Hypertens. 2001;14(9):942–9.
Roerecke M, Kaczorowski J, Tobe SW, Gmel G, Hasan OSM, Rehm J. The effect of a reduction in alcohol consumption on blood pressure: a systematic review and meta-analysis. Lancet Public Health. 2017;2(2):e108–20.
Kraus WE, Bhapkar M, Huffman KM, Pieper CF, Krupa Das S, Redman LM, et al. 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7(9):673–83.
Liang R, Zhang B, Zhao X, Ruan Y, Lian H, Fan Z. Effect of exposure to PM2.5 on blood pressure: a systematic review and meta-analysis. J Hypertens. 2014;32(11):2130–40 discussion 2141.
Intengan HD, Schiffrin EL. Vascular remodeling in hypertension. Hypertension. 2001;38(3):581–7.
Allen AM, Zhuo J, Mendelsohn FAO. Localization and function of angiotensin AT1 receptors. Am J Hypertens. 2000;13(S1):31S–8S.
Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest. 2003;112(1):67–75.
Gkatzionis A, Burgess S. Contextualizing selection bias in Mendelian randomization: how bad is it likely to be? Int J Epidemiol. 2018;48(3):691–701.
Schooling CM, Lopez PM, Yang Z, Zhao JV, Au Yeung SL, Huang JV. Use of Multivariable Mendelian Randomization to Address Biases Due to Competing Risk Before Recruitment. Frontiers in genetics. 2020;11:610852.
Kesteloot H, Decramer M. Age at death from different diseases: the flemish experience during the period 2000-2004. Acta Clin Belg. 2008;63(4):256–61.
Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40(7):597–608.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Westreich D. Berkson’s bias, selection bias, and missing data. Epidemiology. 2012;23(1):159–64.
Wang B, Wu T, Neale MC, Verweij R, Liu G, Su S, et al. Genetic and environmental influences on blood pressure and body mass index in the National Academy of Sciences-National Research Council World War II Veteran Twin Registry. Hypertension. 2020;76(5):1428–34.
Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol. 2017;186(9):1026–34.
Batty GD, Gale CR, Kivimaki M, Deary IJ, Bell S. Comparison of risk factor associations in UK Biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis. BMJ. 2020;368:m131.
Lopez PM, Subramanian SV, Schooling CM. Effect measure modification conceptualized using selection diagrams as mediation by mechanisms of varying population-level relevance. J Clin Epidemiol. 2019;113:123–8.
Zhou B, Bentham J, Di Cesare M, Bixby H, Danaei G, Cowan MJ, et al. Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet. 2017;389(10064):37–55.
Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3(4):524–48.
Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377(1):13–27.
Gapstur SM, Drope JM, Jacobs EJ, Teras LR, McCullough ML, Douglas CE, et al. A blueprint for the primary prevention of cancer: Targeting established, modifiable risk factors. CA Cancer J Clin. 2018;68(6):446–70.
McQueen DV. Strengthening the evidence base for health promotion. Health Promot Int. 2001;16(3):261–8.
Funding
None.
Author information
Authors and Affiliations
Contributions
IIC, MKK and CMS designed the study and interpreted the results. IIC conducted data analysis and wrote the first draft of the manuscript, with critical feedback from MKK and CMS. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
This study only used publicly available data. No original data were collected. Ethical approval for each of the studies included in the investigation can be found in the original publications.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of interest, financial or otherwise.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure S1
. Mendelian randomization estimates of body mass index on total cancer.
Additional file 2: Supplementary Figure S2.
Mendelian randomization estimates of ever-smoking on total cancer.
Additional file 3: Supplementary Figure S3.
Mendelian randomization estimates of systolic (filled) and diastolic (hollow) blood pressure on asthma.
Additional file 4: Supplementary Table S1
. Genetic associations with systolic blood pressure, total and site-specific cancers. Supplementary Table S2. Genetic associations with diastolic blood pressure, total and site-specific cancers. Supplementary Table S3. Power calculations for total and site-specific cancers. Supplementary Table S4. Mendelian randomization estimates of systolic (per 10 mmHg increment) blood pressure on total and site-specific cancers. Supplementary Table S5. Mendelian randomization estimates of diastolic (per 5 mmHg increment) blood pressure on total and site-specific cancers. Supplementary Table S6. Mendelian randomization estimates of systolic (per 10 mmHg increment) and diastolic (per 5 mmHg increment) blood pressure on site-specific cancers in genetic consortia
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chan, I.I., Kwok, M.K. & Schooling, C.M. Blood pressure and risk of cancer: a Mendelian randomization study. BMC Cancer 21, 1338 (2021). https://doi.org/10.1186/s12885-021-09067-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-09067-x