
Abstract
Background
Residual breast cancer after neo-adjuvant chemotherapy (NACT) predicts disease outcome and is a surrogate for survival in aggressive breast cancer (BC) subtypes. Pathological complete response (pCR) rate, however, is lower for luminal B BC in comparison to the triple negative (TNBC) and HER2+ subtypes. The addition of immune checkpoint blockade (ICB) to NACT has the potential to increase pCR rate but is hampered by the lower immunogenicity of luminal B BC. Novel strategies are needed to stimulate the immune response and increase the response rate to ICB in luminal B BC.
Methods
The Neo-CheckRay trial is a randomized phase II trial investigating the impact of stereotactic body radiation therapy (SBRT) to the primary breast tumor in combination with an anti-CD73 (oleclumab) to increase response to anti PD-L1 (durvalumab) and NACT. The trial is designed as a three-arm study: NACT + SBRT +/− durvalumab +/− oleclumab. The result at surgery will be evaluated using the residual cancer burden (RCB) index as the primary endpoint. Six patients will be included in a safety run-in, followed by a randomized phase II trial that will include 136 evaluable patients in 3 arms. Inclusion is limited to luminal B breast cancers that are MammaPrint genomic high risk.
Discussion
combination of ICB with chemotherapy in luminal B BC might benefit from immune priming agents to increase the response rate. As none have been identified so far, this phase II trial will evaluate SBRT and oleclumab as potential immune priming candidates.
Trial registration
trial registered on ClinicalTrials.gov (NCT03875573) on March 14th, 2019.
Similar content being viewed by others


Background
Luminal breast cancer (BC) is characterized by a positive estrogen receptor (ER) status and categorized into two subclasses, A and B [1]. Luminal B BC has a higher tumor cell proliferation rate than luminal A BC, with a breast cancer-specific mortality rate twice as high [1]. The rates of pathological complete response (pCR) with neo-adjuvant chemotherapy (NACT), are poor for luminal B compared to the non-luminal subtypes: 15% in luminal B versus 46% in HER2 positive BC and 45% in triple negative BC (TNBC) [2,3,4]. Luminal B BC patients who don not achieve pCR after NACT have a significantly lower event-free survival, revealing a need to increase response rate to NACT for luminal B BC [5].
Recent developments in immuno-oncology permit classification of tumours according to their immunological contexture: inflamed cancer types, such as melanoma and lung cancer, are characterized by the presence of tumour infiltrating lymphocytes (TILs), high CD8+ T-cell density, and high programmed cell death receptor ligand 1 (PD-L1) positivity of tumour or immune cells, and immune signatures. Because of this, higher overall long-term outcomes with immunotherapy can be attained in inflamed cancer types [6, 7], whereas in non-inflamed cancer types, such as a large proportion of luminal B BC, results with isolated PD-1/PD-L1 blockade are disappointing. Hence, strategies to prime immune responses seem critical to increase clinical benefit of immunotherapy in non-inflamed cancer types [8,9,10]. An area of active research is how to convert non-inflamed cancers into inflamed cancers, leveraging the effects of local and/or systemic treatment and increasing pCR rates with immunotherapy combinations.
The priming of an anti-tumour immune response in early luminal B BC could be attained via a myriad of strategies [11]. In the Neo-CheckRay trial, three priming strategies are used: 1. chemotherapy (given in the three study arms), 2. radiation therapy to the primary BC (given in the three study arms) and 3. blockade of the adenosine pathway (only given in arm 3). The rationale behind these priming strategies will be addressed in the discussion section of the present manuscript.
Methods/design
Study design
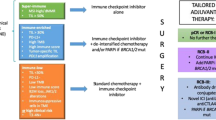
The Neo-CheckRay clinical trial is a multicenter, open-label phase II study that randomizes patients with early stage luminal B BC who are candidate for NACT in a 1:1:1 ratio in 3 arms (Fig. 1):
-
1.
Arm 1: the combination of weekly paclitaxel followed by dose-dense doxorubicin-cyclophosphamide (ddAC) and pre-operative radiation therapy (boost dose) on the primary tumour.
-
2.
Arm 2: treatment of arm 1 with the addition of the anti-PD-L1 antibody durvalumab
-
3.
Arm 3: treatment of arm 2 with the addition of the anti-CD73 antibody oleclumab

Study treatments of the phase II randomised trial. The safety run-in is equivalent to arm 3 of the phase II randomised trial
A safety run-in with inclusion of six patients is planned before starting the randomized phase II part. Those six patients will receive the combination of chemotherapy with durvalumab, oleclumab and SBRT (at the same doses of the IMPs (investigational medicinal product) at identical individual patient timelines than study treatment arm 3).
Objectives and endpoints
Safety run-in objective and endpoints
The objective of the safety run-in is to evaluate the safety and toxicity to perform SBRT (stereotactic body radiation therapy) directed to the primary tumour in combination with durvalumab-oleclumab-paclitaxel and to evaluate the feasibility of performing adequate surgery (breast conserving surgery or mastectomy) without treatment-induced delays. Evaluation of these objectives will be performed by measuring the following endpoints: 1) occurrence of immune related or radiation therapy related toxicity; 2) feasibility of delivering 80% of the total planned dose of paclitaxel and ddAC; 3) feasibility of performing surgery within 6 weeks after the end of the neo-adjuvant treatment. The phase II part of the study will start if all endpoints are met and after approval of an independent data monitoring committee (IDMC) that will review safety data of the 6 patients included in the safety run-in phase. The dosages of the IMPs will be identical in the safety run-in and the phase II randomized trial.
Primary objective and endpoint
The primary objective of the study is to demonstrate an improved tumour response rate after neoadjuvant therapy at surgery with minimal or no residual disease in the primary tumour and in the lymph nodes in arms 2 or 3 versus arm 1. Residual disease will be measured using the residual cancer burden (RCB) index on the surgical specimen. RCB is calculated as a continuous index combining pathologic measurements of the primary tumour (size and cellularity) and lymph node metastases (number and size) [12]. The primary endpoint will be the rate of RCB 0 or 1 at time of surgery. RCB 0 is defined as pCR and RCB 1 is defined as minimal residual disease.
Secondary objectives and endpoints
Secondary objectives are to 1) evaluate the response of the primary tumour irrespective of the response to the pathological lymph nodes; 2) evaluate the response of the pathological lymph nodes irrespective of the response to the primary tumour. These two objectives are of special interest to differentiate the local impact of radiation therapy, which is exclusively directed to the primary cancer and to not to the lymph nodes. Other objectives are to: 3) evaluate the feasibility of performing breast-conserving surgeries in arms 2 and 3 versus arm 1; 4) evaluate invasive disease-free survival (iDFS) in arms 2 and 3 versus arm 1 at 3 years after surgery; 5) evaluate the severity and duration of adverse events in arms 2 and 3 versus arm 1; 6) evaluate the cosmetic changes to the breast in arms 2 and 3 versus arm 1.
Translational objectives and endpoints
The main translational objectives are to 1) evaluate TILs levels in arms 2 and 3 versus arm 1 at different timepoints during the study and correlate these findings to RCB-category [13] and 2) identify potential predictive immune biomarkers by performing a widespread histological and molecular profiling of the pre-, on-, and post-study treatment peripheral blood and tumour tissues, by using immuno- histochemistry and other histological methods such as immunofluorescence, flow cytometry, chromatography and next generation sequencing. Biomarkers include TILs, PD-L1, CD73 and BMI [14] and 3) evaluate the prognostic importance of germinal centers in lymph nodes after treatment and relationship to TILs.
Study population
Inclusion criteria
Main inclusion criteria
-
Histological diagnosis of invasive breast adenocarcinoma that is ER-positive and HER2-negative as per the updated American Society of Clinical Oncology (ASCO) - College of American Pathologists (CAP) guidelines [15] according to local testing and using validated assays in ISO15189-accredited laboratories.
-
ER-positive is defined as having an immunohistochemistry (IHC) of 10% or more and/or and Allred score of 3 or more
-
HER2-negative is defined as having an IHC of 0 or 1+ without in-situ hybridization (ISH) testing OR IHC 2+ and ISH non-amplified with ratio less than 2.0 and if reported, average HER2 copy number < 4 signals/cells OR ISH non-amplified with ratio less than 2.0 and if reported, average HER2 copy number < 4 signals/cells [without IHC]; note: a IHC of 3+ is always considered HER2 positive, independently of the ISH result.
-
-
Luminal B breast tumour, ascertained by a Proliferation Index Ki67 ≥ 15% or histological grade 3.
-
Confirmed MammaPrint genomic high risk.
-
cN0 or cN1 according to TNM classification
-
Absence of distant metastasis (cM0)
-
Multifocal, multicentric unilateral or bilateral breast adenocarcinomas are allowed provided that all foci are ER+/HER2- according to local testing and all foci are able to receive pre-operative radiation therapy treatment within the defined dosimetric constraints.
The complete list of exclusion criteria can be found in the supplementary materials.
Exclusion criteria
Main exclusion criteria:
-
TNM stage cT4 BC, including inflammatory BC
-
Presence of any distant metastasis
The complete list of exclusion criteria can be found in the supplementary materials.
Treatments and interventions
Treatments
The chemotherapy backbone of the trial consists of 12 administrations of intravenous (IV) weekly paclitaxel (80 mg/m2) followed by 4 administrations of doxorubicin (60 mg/m2) plus cyclophosphamide (600 mg/m2 IV) in a dose dense scheme (ddAC), i.e. every 2 weeks (q2w) (Table 1). Durvalumab (anti PD-L1) is given q4w 1500 mg IV. Oleclumab (anti-CD-73; MEDI9447) is given q2w for 5 administrations, then q4w during 2 administrations (3000 mg IV). A detailed overview can be found in the study scheme (Fig. 1).
Pre-operative radiation therapy to the primary tumour will be given at a dose of 24 Gy in 3 fractions using an SBRT technique with the 3rd fraction given on the same day as the 5th paclitaxel, 2nd durvalumab and 3rd oleclumab administration (Fig. 1). Pathological lymph nodes will not be irradiated. Special care will be taken to reduce the dose to the axillary lymph nodes, the heart, the lungs and the skin. To allow for optimal image guidance during SBRT treatment, at least 3 MRI-compatible markers must be placed in the primary tumour during the screening period. Image guidance during treatment will be performed using daily CBCT (cone beam computed tomography) and markers-based matching. MRI in supine position, preferably in radiation therapy treatment position, is recommended to facilitate contouring.
Surgery should be planned 2–6 weeks after completion of ddAC and will consist of breast conserving surgery or mastectomy and a sentinel node procedure and/or an axillary lymph node dissection.
Interventions
The study design (Fig. 1) charts the timing of all scheduled blood samplings, biopsies and tumour imaging procedures. A new biopsy of the primary tumour is planned 1 week after ending the radiation therapy (i.e. week 6) to evaluate tumour and tumour microenvironment changes induced by the first 5 weeks of paclitaxel, durvalumab, oleclumab and radiation therapy. An MRI is planned 12 weeks after the start of study treatment, before switching the chemotherapy to ddAC. Blood samples for translational research are scheduled throughout the pre-operative phase.
Treatments after surgery
Adjuvant endocrine therapy is given according to standard of care (SOC). Post-operative radiation therapy will be administered according to SOC, with the only exception that no boost to the tumour bed is allowed, because the pre-operative radiation therapy to the primary tumour is to be considered as an anticipated boost.
Statistical analysis
Sample size
Six patients will be included in the safety run-in. In the phase II part, patients will be randomized in a 1:1:1 ratio between 3 arms and the primary endpoint that will be measured on those patients is the binary achievement of RCB 0–1 versus RCB 2–3. The trial is designed to compare arm 2 to arm 1 as well as arm 3 to arm 1. The trial is not powered to compare arms 2 and 3 and no specific hypothesis is formulated for that comparison. It is expected that arm 1 will achieve an RCB 0–1 rate of 15% and the experimental treatment arms will be considered of interest for further investigation if this rate can be increased to 45%. This estimation for arms 2 and 3 are based on the first results of the I-SPY 2 trial (2017): the addition of immune checkpoint blockade (ICB) to standard NACT increased pCR from 13.6 to 34.2% [16]. Taking into account the addition of radiation therapy and the use of RCB 0–1 as endpoint, we hypothesize an increase in RCB 0–1 rates from 15 to 45% in either arm 2 or 3. There will be then two hypotheses testing, both aiming to reject the null hypothesis of equality of the RCB 0–1 rates between the control and in the experimental arms. A two-sided alpha level of 2.5% will be used and a power of 80% has been targeted in case the true RCB rate in either arm 2 or arm 3 is at least 45%. The alpha level of 2.5% was chosen to adjust for multiplicity. No continuity correction was used, and a pooled estimate of variance was chosen to estimate requested sample size. Sample size calculations are summarized in Table 2. With those assumptions, 44 evaluable patients per arm are needed, i.e. a total of 132 patients. In order to take into account a 5% non-evaluability rate and a 5% non-eligibility rate, this sample size will be increased to 147 patients to be randomized. Assuming 20% screening failures (taking into account MammaPrint patients that are screened as MammaPrint genomic low risk), 184 patients will have to be screened.
Data analysis
Primary endpoint analysis will be carried out on the eligible and randomized patients who have been operated and in whom the residual cancer burden was measured. The observed proportions will be compared by a chi square test without continuity correction at an alpha two-sided level of 2.5%. Confidence intervals for the difference between proportions will be provided at the usual 95% level. The comparison of RCB 0–1 rates between arm 2 and arm 1 as well as arm 3 and arm 1 will be performed. As exploratory analysis, a confidence interval for the difference between proportions in arm 2 and arm 3 will be provided.
Discussion
Approximately 40% of patients with early-stage luminal B BC who receive NACT experience recurrent disease within 5 years [5], therefore new treatment strategies to decrease recurrence rates are greatly needed. In recent years, an emerging treatment strategy is the use of ICB. Early trials of single-agent ICB in BC show evidence of a modest response, less than in some other cancers due to the low baseline immunogenicity of most BCs [17]. Recent advances in the field of immunogenomics identified different immune subtypes that are hypothesized to define how immune response patterns impact prognosis [18]. In BC, the immune response of hormone receptor positive (luminal) BC is lower compared to the TNBC and HER2+ subtypes, assumedly because luminal BC is generally less inflamed, characterized by a lower presence of TILs and lower PD-L1 expression [19]. In TNBC and HER2+ BC, increased TIL concentration is correlated with response to chemotherapy and immunotherapy, and is associated with increased survival, whereas in luminal BC the prognostic and predictive value of increased TIL concentration is not fully established [7].
An attractive strategy to increase the benefit of immunotherapy in luminal B BC seems to be to prime the immune response using a combination strategy with chemotherapy, a doublet immunotherapy, radiation therapy or other agents. The I-SPY2 trial investigated the addition of pembrolizumab (anti PD-1) to a standard NACT backbone in TNBC and luminal B BCs [20]. The addition of pembrolizumab increased pCR rates from 13 to 30% in luminal B BC and from 22 to 60% in TNBC. These results indicate that non-inflamed” tumours might benefit from immunotherapy when combined with other treatments, such as chemotherapy. In TNBC, the phase III trial KEYNOTE 522 demonstrated superiority of the pembrolizumab-NACT combination with 64.8% pCR and 51.2% pCR in the NACT-only group [21], whereas for luminal B BC, KEYNOTE 756 is ongoing.
An active domain of research is the identification of non-chemotherapeutic agents able to prime the immune response and to further enhance the conversion to more inflamed tumours with the hope of increasing response rates to immunotherapy. In the Neo-CheckRay trial, radiation therapy to the primary tumour and the use of an anti-CD-73 are investigated as potential strategies to increase the response rate following immunotherapy-chemotherapy combinations.
In recent years, a large amount of clinical trials and animal studies have described the synergistic effects on local and distant tumour control of combining radiation therapy with immunotherapy [22,23,24,25,26]. In BC models, radiation has been shown to induce T cell priming via antigenic release and by activation of the innate immunity [22, 27, 28]. Pivotal pre-clinical work by Vanpouille-Box et al. showed that a fractionation schedule of three fractions of 8 Gy given daily induces a better activation of the cGAS-STING pathway in comparison to treatments with a higher dose per fraction [29]. Despite the fact that the 3 × 8 Gy schedule has been used in several clinical trials to evaluate its efficacy and toxicity in combination with immunotherapy [30,31,32], this fractionation schedule remains yet to be validated. While an excessively high radiation fraction dose might suppress the immune response or increase toxicity, an insufficient dose might not induce an immune response at all. Apart from dose/fractionation parameters, also the sequencing of radiation therapy when combined with ICB remains largely unknown, further highlighting the importance of the need for more clinical trials. A challenge of delivering pre-operative radiation therapy in combination with immunotherapy is the concern of post-operative toxicities, with potential impacts on breast cosmesis, lymphedema, fibrosis and surgical outcomes, especially in case of breast reconstruction. The impact on these parameters can be estimated from other pre-operative trials combing radiation therapy with chemotherapy [33], but it remains unknown how the addition of immunotherapy will modulate these effects. For this, breast cosmesis will be monitored during and after the Neo-Checkray trial, using standardised breast photography and clinical examinations.
Oleclumab is a human immunoglobulin monoclonal antibody (mAb) that selectively binds to and inhibits the ectonucleotidase activity of CD73. CD73 converts adenosine monophosphate (AMP) into adenosine. Adenosine presents anti-inflammatory and immunosuppressive effects, mediated by the adenosine receptors expressed on immune cells [34, 35]. CD73 therefore serves as an immune checkpoint, with the clinical association between elevated CD73 expression and poor prognosis being well documented in several tumour types [36,37,38]. The therapeutic potential of blocking adenosine pathways with monoclonal antibodies targeting is currently being tested in phase II trials [39].
Radiation therapy has pre-clinically been shown to act synergistically with anti-CD-73 through the prevention of adenosine-mediated immunosuppression [40]. Recent research suggests that CD73 may be a radiation-induced checkpoint, and that CD73 blockade in combination with radiation therapy and ICB might improve patient response to therapy [41]. Pre-clinical data show that the combination of CD73 inhibition and radiation therapy has the following effect: 1) enhancement of the radiation-induced activation of the antitumour immune response, 2) restriction of the immunosuppressive action of CD39/CD73 on circulating immune cells and 3) attenuation of adverse late effects of radiation therapy through inhibition of fibrosis [41,42,43].
The Neo-CheckRay trial was designed to harness these potential synergies, by combining durvalumab with oleclumab and radiation therapy to the primary BC in addition to a standard chemotherapy backbone. Parts of this combination have been tested for safety, such as the combination of radiation therapy and docetaxel [33], radiation therapy with PD-L1 blockade [32], and the combination of oleclumab, durvalumab and paclitaxel [44]. The combination of all 4 treatment agents will be tested in a safety run-in phase of the Neo-CheckRay trial before proceeding to the randomized phase II trial.
Given the choice of RCB 0–1 as primary endpoint of the current trial, it was decided to deliver radiation therapy to the three treatment arms to avoid an imbalance in local treatment between the study arms. Indeed, radiation therapy might increase the local response without necessarily reflecting an impact on the systemic response and is therefore more difficult to discriminate as being an independent surrogate of long-term outcome. Moreover, our study design allows singling out the effect of durvalumab and oleclumab without the results being disbalanced by radiation therapy. The drawback of this choice is that arm 1 is not a commonly accepted standard of care, which would have been chemotherapy alone, excluding the individual impact of pre-operative SBRT.
A similar trial could have been designed with the systemic treatments in the adjuvant setting or in the metastatic setting. The neo-adjuvant setting, however, with a primary tumour in place, presents unique advantages for priming the anti-tumour immune response and potential eradication of micrometastatic disease [45]. Pre-clinical models of BC support the superiority of neo-adjuvant immunotherapy compared to adjuvant immunotherapy [46]. With the intact primary cancer still in place, radiation therapy and ICB might help to convert the tumour into an in-situ, individualized vaccine; hypothetically preventing patients against metastasis [47]. In addition, neo-adjuvant treatment followed by complete surgical resection of the tumour allows a rapid and thorough assessment of response with the possibility of performing a wide range of translational research on the surgical specimen.
The Neo-Checkray trial targets a subselection of luminal B cancers by strictly including patients with a MammaPrint test showing a genomic high risk of relapse. Patients with luminal B BC and a genomic high risk of relapse derive a greater benefit from chemotherapy than patients with a genomic low risk result [48]. The use of this test ensures that patients included in the trial derive benefit of chemotherapy. Unlike with chemotherapy, biomarkers to select patients for immunotherapy have not yet been identified for BC patients. Hence, an important part of the translational research objectives of the Neo-CheckRay study is to evaluate the predictive value of potential biomarkers, such as PD-L1 and TILs, and to identify other biomarkers, both in situ as well as in the systemic circulation [49], that might help to better tailor therapy in this setting.
Few other trials are investigating the role of pre-operative radiation therapy in combination with immunotherapy in BC. A phase I clinical trial (NCT03366844 - recruiting) is evaluating the safety of 3 × 8 Gy radiation therapy to the primary BC in combination with pembrolizumab for TNBC and ER + HER2- BC, without the use of chemotherapy [50]. Another pre-operative design in luminal BC is the CBCV trial (NCT03804944 – not yet recruiting), a 4 arms study consisting of 1) radiation therapy 3 × 8 Gy (RT) + hormonal treatment (HT); 2) RT/HT + pembrolizumab; 3) RT/HT + CDX-301 (anti-CD135); 4) RT/HT + pembrolizumab + CDX-301 [51]. In TNBC, the PANDoRA trial (NCT03872505 – not yet recruiting) will evaluate the addition of radiation therapy (3x8Gy) to non-anthracycline chemotherapy and durvalumab in TNBC [52]. To our knowledge, the Neo-CheckRay trial is the first trial to examine the effect of the pre-operative addition of radiation therapy to the combination of immunotherapy with standard NACT in early luminal B BC.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analysed yet during the current study.
Abbreviations
- AMP:
-
Adenosine monophosphate
- BC:
-
Breast cancer
- CBCT:
-
Cone beam computed tomography
- ddAC:
-
dose-dense doxorubicin-cyclophosphamide
- ER:
-
Estrogen receptor
- HT:
-
Hormonal treatment
- ICB:
-
Immune checkpoint blockade
- iDFS:
-
invasive disease-free survival
- IDMC:
-
Independent data monitoring committee
- IHC:
-
Immunohistochemistry
- IMP:
-
Investigational medicinal product
- ISH:
-
In-situ hybridization
- IV:
-
Intravenous
- mAb:
-
monoclonal antibody
- NACT:
-
Neo-adjuvant chemotherapy
- pCR:
-
pathological complete response
- PD-L1:
-
Programmed cell death receptor ligand 1
- RCB:
-
Residual cancer burden
- SBRT:
-
Stereotactic body radiation therapy
- SOC:
-
Standard of care
- TIL:
-
Tumor infiltrating lymphocyte
- TNBC:
-
Triple negative breast cancer
References
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. https://doi.org/10.1073/pnas.191367098s.
Bonnefoi H, Litière S, Piccart M, MacGrogan G, Fumoleau P, Brain E, et al. Pathological complete response after neoadjuvant chemotherapy is an independent predictive factor irrespective of simplified breast cancer intrinsic subtypes: a landmark and two-step approach analyses from the EORTC 10994/BIG 1-00 phase III trial. Ann Oncol. 014;25(6):1128–36. https://doi.org/10.1093/annonc/mdu118.
Gianni L, Pienkowski T, Im YH, Roman L, Tseng LM, Liu MC, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(1):25–32. https://doi.org/10.1016/S1470-2045(11)70336-9.
Poggio F, Bruzzone M, Ceppi M, Pondé NF, la Valle G, del Mastro L, et al. Platinum-based neoadjuvant chemotherapy in triple-negative breast cancer: a systematic review and meta-analysis. Ann Oncol. 2018;29(7):1497–508. https://doi.org/10.1093/annonc/mdy127.
Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–72. https://doi.org/10.1016/S0140-6736(13)62422-8.
T. F. Gajewski, L. Corrales, J. Williams, B. Horton, A. Sivan, and S. Spranger, “Cancer Immunotherapy Targets Based on Understanding the T Cell-Inflamed Versus Non-T Cell-Inflamed Tumor Microenvironment BT - Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy,” P. Kalinski, Ed. Cham: Springer International Publishing, 2017, pp. 19–31.
Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19(1):40–50. https://doi.org/10.1016/S1470-2045(17)30904-X.
Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, et al. Safety and antitumor activity of pembrolizumab in patients with estrogen receptor–positive/human epidermal growth factor receptor 2–negative advanced breast cancer. Clin Cancer Res. 2018;24(12):2804–11. https://doi.org/10.1158/1078-0432.CCR-17-3452.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. https://doi.org/10.1038/nature21349.
Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN solid tumor study. Breast Cancer Res Treat. 2017;167(3):1–16. https://doi.org/10.1007/s10549-017-4537-5.
Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;7807(September) in press. https://doi.org/10.1126/scitranslmed.aat7807.
Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol. 2007;25(28):4414–22. https://doi.org/10.1200/JCO.2007.10.6823.
Luen SJ, Salgado R, Dieci MV, Vingiani A, Curigliano G, Gould RE, et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy original article. Cerebrospinal fluid cell-free tumour DNA as a Liq biopsy brain tumours. 2019;30(2):236–42. https://doi.org/10.1093/annonc/mdy547.
Floris G, et al. Body mass index and tumor infiltrating lymphocytes in triple-negative breast cancer. JNCI J Natl Cancer Inst. 2020. https://doi.org/10.1093/jnci/djaa090.
Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL, et al. Estrogen and progesterone receptor testing in breast cancer: American society of clinical oncology/college of American pathologists guideline update. Arch Pathol Lab Med. 2020;144(5):545–63. https://doi.org/10.5858/arpa.2019-0904-SA.
R. Nanda and M. Liu, “Nanda R, Liu MC, Yau C, et al. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): Results from I-SPY 2.,” J Clin Oncol, 35, suppl; abstr 506, 2017.
Luen S, Virassamy B, Savas P, Salgado R, Loi S. The genomic landscape of breast cancer and its interaction with host immunity. Breast. Oct. 2016;29:241–50. https://doi.org/10.1016/j.breast.2016.07.015.
Thorsson V, et al. The Immune Landscape of Cancer. Immunity. 2018;48(4):812–830.e14. https://doi.org/10.1016/j.immuni.2018.03.023.
Stanton SE, Adams S, Disis ML. Variation in the incidence and magnitude of tumor-infiltrating lymphocytes in breast Cancer subtypes: a systematic review. JAMA Oncol. 2016;2(10):1354–60. https://doi.org/10.1001/jamaoncol.2016.1061.
Nanda R, et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer. JAMA Oncol. 2020;60637. https://doi.org/10.1001/jamaoncol.2019.6650.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–21. https://doi.org/10.1056/NEJMoa1910549.
Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015;16(13):e498–509. https://doi.org/10.1016/S1470-2045(15)00007-8.
Pilones KA, Vanpouille-Box C, Demaria S. Combination of radiotherapy and immune checkpoint inhibitors. Semin Radiat Oncol. 2015;25(1):28–33. https://doi.org/10.1016/j.semradonc.2014.07.004.
Derer A, Frey B, Fietkau R, Gaipl US. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol Immunother. 2016;65(7):779–86. https://doi.org/10.1007/s00262-015-1771-8.
Vatner RE, Cooper BT, Vanpouille-Box C, Demaria S, Formenti SC. Combinations of Immunotherapy and Radiation in Cancer Therapy. Front Oncol. 2014;4. https://doi.org/10.3389/fonc.2014.00325.
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18(5):313–22. https://doi.org/10.1038/nrc.2018.6.
Garnett CT, Palena C, Chakarborty M, Tsang K, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64(21):7985–94. https://doi.org/10.1158/0008-5472.CAN-04-1525.
Filatenkov A, Baker J, Mueller AMS, Kenkel J, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. 2015;21(16):3727–39. https://doi.org/10.1158/1078-0432.CCR-14-2824.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8(1):15618. https://doi.org/10.1038/ncomms15618.
Demaria S, Romano E, Brackstone M, Formenti SC. Immune induction strategies to enhance responses to PD-1 blockade: lessons from the TONIC trial. J Immunother Cancer. 2019;7(1):1–4. https://doi.org/10.1186/s40425-019-0783-x.
Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of Pembrolizumab after stereotactic body radiotherapy vs Pembrolizumab alone on tumor response in patients with advanced non–small cell lung Cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol. 2019;5(9):1276–82. https://doi.org/10.1001/jamaoncol.2019.1478.
Luke JJ, et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients With Advanced Solid Tumors. J Clin Oncol. 2018:JCO.2017.76.2229. https://doi.org/10.1200/JCO.2017.76.2229.
Bondiau P-Y, Courdi A, Bahadoran P, Chamorey E, Queille-Roussel C, Lallement M, et al. Phase 1 clinical trial of stereotactic body radiation therapy concomitant with neoadjuvant chemotherapy for breast cancer. Int J Radiat Oncol Biol Phys. 2013;85(5):1193–9. https://doi.org/10.1016/j.ijrobp.2012.10.034.
Ohta A, et al. A2A adenosine receptor protects tumors from antitumor T cells. PNAS. 2006.
Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. 2014;134(6):1466–73. https://doi.org/10.1002/ijc.28456.
Yu M, et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat Commun. 2020;11(1). https://doi.org/10.1038/s41467-019-14060-x.
Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res. 2016;22(1):158–66. https://doi.org/10.1158/1078-0432.CCR-15-1181.
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107(4):1547–52. https://doi.org/10.1073/pnas.0908801107.
L. Buisseret, “Paclitaxel + Carboplatin + Durvalumab With or Without Oleclumab for Previously Untreated Locally Recurrent Inoperable or Metastatic TNBC (SYNERGY) NCT03616886,” clinicaltrials.gov. [Online]. Available: https://clinicaltrials.gov/ct2/show/NCT03616886. [Accessed: 24-May-2020].
Vaupel P, Multhoff G. Adenosine can thwart antitumor immune responses elicited by radiotherapy Therapeutic strategies alleviating protumor ADO activities. Strahlenther Onkol. 2016;192(5):279–87. https://doi.org/10.1007/s00066-016-0948-1.
Wennerberg E, et al. CD73 blockade promotes dendritic cell infiltration of irradiated tumors and tumor rejection. Cancer Immunol Res. 2020. https://doi.org/10.1158/2326-6066.CIR-19-0449.
de Leve S, et al. Loss of CD73 prevents accumulation of alternatively activated macrophages and the formation of prefibrotic macrophage clusters in irradiated lungs. FASEB J. 2017;31(7):2869–80. https://doi.org/10.1096/fj.201601228R.
Wirsdorfer F, et al. Extracellular adenosine production by ecto-50-nucleotidase (CD73) enhances radiation-induced lung fibrosis. Cancer Res. 2016;76(10):3045–56. https://doi.org/10.1158/0008-5472.CAN-15-2310.
L. Buisseret, “Paclitaxel + Carboplatin + Durvalumab With or Without Oleclumab for Previously Untreated Locally Recurrent Inoperable or Metastatic TNBC (SYNERGY) NCT03616886,” clinicaltrials.gov.
Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020;367(6477):eaax0182. https://doi.org/10.1126/science.aax0182.
Liu J, Blake SJ, Yong MCR, Harjunpää H, Ngiow SF, Takeda K, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6(12):1382–99. https://doi.org/10.1158/2159-8290.CD-16-0577.
Jatoi I, Benson JR, Kunkler I. Hypothesis: can the abscopal effect explain the impact of adjuvant radiotherapy on breast cancer mortality? npj Breast Cancer. 2018;4(1):8. https://doi.org/10.1038/s41523-018-0061-y.
Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70-gene signature as an aid to treatment decisions in early-stage breast Cancer. N Engl J Med. 2016;375(8):717–29. https://doi.org/10.1056/NEJMoa1602253.
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24(12):1845–51. https://doi.org/10.1038/s41591-018-0232-2.
“Breast Cancer Study of Preoperative Pembrolizumab + Radiation,” clinicaltrials.gov (NCT03366844). Accessed on June 1st 2020. .
“CBCV: Converting HR+ Breast Cancer Into an Individualized Vaccine,” clinicaltrials.gov. NCT03804944. Accesed June 1, 2020. .
“PANDORA: Pre-operAtive Non-Anthracycline Chemotherapy, Durvalumab +/− RAdiation Therapy in Triple Negative Breast Cancer,” ClinicalTrials.gov. NCT03872505. Accessed 1 April 2020. .
Acknowledgements
Not applicable.
Trial information
Sponsor identification
Sponsor: Institut Jules Bordet, Rue Héger Bordet 1, 1000 Brussels, Belgium.
Sponsor Protocol Number: IJB-LBC-NEOCHECKRAY-2018.
Trial identification
Trial registered on ClinicalTrials.gov (NCT03875573) on March 14th, 2019; accessible at https://clinicaltrials.gov/ct2/show/NCT03875573.
EudraCT Number: 2018–004165-13.
Role of study sponsor and funders
The sponsor (Institut Jules Bordet, Rue Héger Bordet 1, Brussels) detains ultimate authority on study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication.
Composition, roles, and responsibilities of the coordinating centre, steering committee, endpoint adjudication committee, data management team
The coordinating centre is the clinical trials support unit (CTSU) of the Jules Bordet Institute. Information on the composition, roles and responsibilities of the CTSU can be found at https://ctsu.bordet.be/#.
Protocol version and date on which this manuscript was based
Protocol version 2, dated 01/02/2020.
Names, affiliations, and roles of protocol contributors
Alex De Caluwé, Institut Jules Bordet, radiation oncologist, study chair.
Laurence Buisseret, Institut Jules Bordet, medical oncologist.
Philip Poortmans, GZA Antwerp, radiation oncologist.
Roberto Salgado, GZA Antwerp, pathologist.
Marianne Paesmans, Institut Jules Bordet, study statistician.
Fabien Reyal, Institut Curie, surgeon.
Martine Piccart, Institut Jules Bordet, medical oncologist.
Emanuela Romano, Institut Curie, medical oncologist, study co-chair.
Michail Ignatiadis, medical oncologist.
Study sites
Safety run-in: Institut Jules Bordet, Brussels, Belgium.
Phase II randomized trial: Institut Jules Bordet, Brussels, Belgium; Institut Curie, Paris, France; 4 other sites in Belgium and France to be confirmed.
Biological specimens
Biological specimens will be collected and stored for molecular analysis.
Protocol amendments
Important protocol modifications will be communicated to investigators, research ethics review process, regulators and trial registries.
Funding
This study is supported by AstraZeneca and drug supply for oleclumab and durvalumab is provided by AstraZeneca. The trial is an investigator initiated academic trial in which AstraZeneca has no role in the design of the study, in the collection, analysis and interpretation of the data, and in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Study design: ADC, LB, PP, MP, ER, MI. Manuscript writing: ADC, LB, PP, DVG, RS, CS, DL, MP, LC, DS, CV, FR, VI, DE, MP, ER, MI. Study conduct and implementation: ADC, LB, PP, DVG, RS, CS, DL, MP, LC, DS, CV, FR, VI, DE, MP, ER, MI. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Clinical research was performed in accordance with the Declaration of Helsinki and the protocol was approved by ethical committees in Belgium and France. In Belgium: Commissie Medische Ethiek UZ Brussels/VUB, Laarbeeklaan 101, 1090 Brussels, Reference number: 2019/P/02. In France: Ethics committee: CHU de Grenoble, Comité de Protection des personnes, CS 10217 38043, Grenoble Cedex 9, Reference number: 08. Réf. CPP: 20-JUBO-01.
Informed consent to participate in the study will be obtained from all participants.
Consent for publication
This present manuscript does not contain any patient data because only the trial protocol is described, hence consent for current publication is not applicable. The patients that are currently being included in the trial provide written consent for publication of trial results after reading and understanding an informed consent form.
Competing interests
ADC: institutional research grant from AstraZeneca. LB: institutional research grant from AstraZeneca; speaker honoraria from BMS; travel grant from Roche. PP: part-time medical advisor for Sordina IORT Technologies s.p.a., since 1 April 2020, not related to this work. DVG: Advisory board/Honoraria received: Sanofi, Accuray, Merck-Pfizer, Takeda and Novartis. RS: non-financial support from Merck, non-financial support from BMS, other from Puma Biotechnology, other from Roche, other from Roche, other from Merck. DE: Research Funding (ESMO Fellowship): Novartis. Speaker fee: Janssen. MP: Board Member (Scientific Board): Oncolytics; Consultant (honoraria): AstraZeneca, Camel-IDS, Crescendo Biologics, Debiopharm, G1 Therapeutics, Genentech, Huya, Immunomedics, Lilly, Menarini, MSD, Novartis, Odonate, Periphagen, Pfizer, Roche, Seattle Genetics. Research grants to my Institute: AstraZeneca, Lilly, MSD, Novartis, Pfizer, Radius, Roche-Genetech, Servier, Synthon. No stock ownership. ER: no disclosures relevant to this project. MI: Consultant or advisory role (honoraria): Celgene, Novartis, Pfizer, Seattle Genetics, Tesaro. Research grants to my Institute: Roche, Menarini Silicon Biosystems, Janssen Diagnostics, Pfizer. No stock ownership. Travel grants: Pfizer, Amgen. All other authors: no competing interests disclosed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
De Caluwé, A., Buisseret, L., Poortmans, P. et al. Neo-CheckRay: radiation therapy and adenosine pathway blockade to increase benefit of immuno-chemotherapy in early stage luminal B breast cancer, a randomized phase II trial. BMC Cancer 21, 899 (2021). https://doi.org/10.1186/s12885-021-08601-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08601-1