
Abstract
Background
Lung adenocarcinoma (LUAD) is the most common pathology subtype of lung cancer. In recent years, immunotherapy, targeted therapy and chemotherapeutics conferred a certain curative effects. However, the effect and prognosis of LUAD patients are different, and the efficacy of existing LUAD risk prediction models is unsatisfactory.
Methods
The Cancer Genome Atlas (TCGA) LUAD dataset was downloaded. The differentially expressed immune genes (DEIGs) were analyzed with edgeR and DESeq2. The prognostic DEIGs were identified by COX regression. Protein-protein interaction (PPI) network was inferred by STRING using prognostic DEIGs with p value< 0.05. The prognostic model based on DEIGs was established using Lasso regression. Immunohistochemistry was used to assess the expression of FERMT2, FKBP3, SMAD9, GATA2, and ITIH4 in 30 cases of LUAD tissues.
Results
In total,1654 DEIGs were identified, of which 436 genes were prognostic. Gene functional enrichment analysis indicated that the DEIGs were involved in inflammatory pathways. We constructed 4 models using DEIGs. Finally, model 4, which was constructed using the 436 DEIGs performed the best in prognostic predictions, the receiver operating characteristic curve (ROC) was 0.824 for 3 years, 0.838 for 5 years, 0.834 for 10 years. High levels of FERMT2, FKBP3 and low levels of SMAD9, GATA2, ITIH4 expression are related to the poor overall survival in LUAD (p < 0.05). The prognostic model based on DEIGs reflected infiltration by immune cells.
Conclusions
In our study, we built an optimal prognostic signature for LUAD using DEIGs and verified the expression of selected genes in LUAD. Our result suggests immune signature can be harnessed to obtain prognostic insights.
Similar content being viewed by others


Introduction
Lung cancer is one of the most common diseases with the highest morbidity and mortality, in which the lung adenocarcinoma accounts for 40% of all cases. In recent years, the morbidity and mortality of lung adenocarcinoma have gradually increased [1]. Chemotherapy, radiotherapy and targeted therapy are the most common therapeutic methods for advanced lung adenocarcinoma. Although multiple therapeutics have been used in LUAD, the overall effective rate is unsatisfactory.
Increasing evidence suggested that tumor microenvironment (TME) which is composed of tumor cells, immune cells, stromal cells, inflammatory mediators and extracellular matrix [2], taking part in the tumor progression and drug resistance [3, 4]. Among them, immune cells and inflammatory mediators have been proved to be valuable for the prognostic of LUAD [5]. Much attention has been paid on the immune microenvironment of LUAD.
Current studies showed that immunology and immunogenomics were closely tied to the development of LUAD [6, 7]. Immunotherapy is expected to replace the traditional treatment based on a number of clinical studies. In recent years, the emergence of immune checkpoint inhibitors has enabled a dramatic progress in cancer treatment [8, 9]. How to select the patients who really benefit from immunotherapy has become an urgent problem to be solved. It is important to identify biomarkers that can predict disease prognosis and identify the patients who have the greatest curative effect. S-PD-L1 and T-PD-1 were verified as the independent prognostic factors for non small-cell lung cancer (NSCLC) patients by Paulsen [10]. Their combination added significant prognostic impact within each pathologic stage. Several studies suggested that tumor mutational burden (TMB) [11, 12], mismatch repair (MMR) [13, 14] are new biomarkers for prediction of response to PD-L1 treatment. However, cause of the heterogeneity, accurate theranostic biomarkers are still lacking. The exploration of biomarkers in the immune microenvironment remains largely unknown. In this study, we combined multiple datasets from TCGA LUAD to develop and validate a prognosis prediction model for LUAD. Meanwhile, an optimal prognostic model with the identified DEIGs via lasso regression was established by us. Our aim is to give a more in-depth view of the prognostic potential of DEIGs in clinical and provides a foundation for future, in-depth immune-related work of LUAD.
Materials and methods
All methods were carried out in accordance with relevant guidelines and regulations.
Data preprocessing
TCGA LUAD dataset legacy-archive (hg19) was downloaded from NCI’s Genomic Data Commons (GDC) (https://portal.gdc.cancer.gov) using R package ‘TCGA biolinks’ [15], and only “Primary solid Tumor” and “Solid Tissue Normal” samples were included. Furthermore, the immune-related genes were derived from InnateDB (https://www.innatedb.com) [16]. While the estimated infiltration abundance of immune cells of LUAD samples were obtained by TIMER (https://cistrome.shinyapps.io/timer/) [17].TIMER is a resource providing pre-calculated levels of six tumor-infiltrating immune subsets for 10,897 tumors from 32 cancer types.
Identification of prognostic DEIGs
Differentially expressed RNAs were detected using DESeq2 [18] and edgeR [19]. RNAs with ‘|log2 (fold change) | > 1’, ‘p value < 0.05’ and ‘fdr < 0.3’ in both methods were considered to be differentially expressed. COX regression was employed to identify prognostic DEIGs.
Annotation of prognostic DEIGs
The R package ‘ClusterProfiler’ [20] was employed for pathway enrichment analysis with DEIGs. Functional enrichment analyses, via the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [21], were conducted to explore potential molecular mechanisms of the differentially expressed prognostic DEIGs.
PPI network construction and hub-genes identification
PPI network was inferred by STRING using the prognostic differentially expressed immune genes with p value< 0.05 in cox test [22]. Hub genes were identified by cytoscape.
Modeling via lasso regression
We used glmnet package to fit regularized Cox models. The function cv.glmnet was used to compute K-fold cross-validation (CV) for the Cox model with parameters ‘ family=“cox”, nfolds=10’. The optimal λ value and a cross validated error plot were shown as below. The left vertical line indicated where the CV-error curve hits its minimum. And the right vertical line showed the most regularized model with CV-error within 1 standard deviation of the minimum. We then extracted the lambda.min for model construction.
The whole TCGA dataset was divided into 70% of training samples and 30% of test samples. The prediction model was built on the most frequent gene set with effective coefficients in the lasso regression using R package ‘glmnet’ [23] for 1000 iterations on the training dataset. The risk score was defined as the sum of the normalized expression of genes multiplied by their coefficients in the gene set. ROC was used to evaluate the cutoff of risk scores as a predicting factor for the survival of LUAD patients at 5 years prior to death. After dividing the patient into two groups according to the risk score, ‘Survminer’ was employed for survival analysis for both training and testing data. The pearson correlation coefficients of risk score and immune cells/immune cells markers were calculated by the R package ‘ggpubr’.
Immunohistochemistry
This study recruited 30 patients of LUAD getting surgery at Tumor Hospital of Shaanxi province between January 2014 and December 2015 whom had no prior chemotherapy or radiotherapy. Antibodies included a rabbit polyclonal anti-FERMT2 antibody at a dilution of 1:50, anti- FKBP3 antibody at a dilution of 1:50, (all from Proteintech Group, China), anti-SMAD9 antibody at a dilution of 1:100, anti-GATA2 antibody at a dilution of 1:100, anti-ITIH4 antibody at a dilution of 1:50 (all from Beijing Biosynthesis Biotechnology, China). PBS was used to displace the primary antibody as the negative control. The histological diagnosis was performed by 3 independent, experienced pathologists for all the cases. The Immunohistochemistry (IHC) was performed according to our previous study [24]. Five micrometer-thick sections were cut from the human lung adenocarcinoma tissue and fixed in 10% buffered formalin overnight and paraffin-embedded. The slides were deparaffinized and rehydrated in graded alcohols, followed by antigen retrieval in a microwave oven. Slides were blocked with 10% normal goat serum for 20 min at 37 °C to reduce nonspecific binding. The slides were incubated overnight at 4 °C [25]. After being washed, Horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG was used as secondary antibody, and then visualized with 3,3′-diaminobenzidine (DAB) solution. Finally, hematoxylin was used to counterstain the section. The percentage of positive cells was classified into 5 score ranges: < 10% (0),10 to 25% (1), 25 to 50% (2), 50 to 75% (3), and > 75% (4). The intensity of staining was divided into 4 groups: no staining (0), light brown (1), brown (2), and dark brown (3). The staining positivity was determined using immunoreactivity score (IRS) which is the product of intensity score and quantity score. An overall score of > 6 as strong positive, > 3 as weak positive, and ≤ 3 was defined as negative.
Results
Identification of prognostic DEIGs
The immune-related genes were downloaded from InnateDB. The differentially expressed gene analysis was performed by edgeR and DESeq2, and only DEIGs detected by both methods were included. Four hundred thirty-six genes were identified with p value <= 0.05 in cox tests by the R package ‘survival’ (Table S1).
PPI network and hub genes
To gain insights into the core pathways exerted by those DEIGs, we constructed PPI network and identified core modules within the network. PPI analysis demonstrated that FANCI, MAD2L1, ECT2, PLK4, PCNA, BUB1B, RACGAP1, PRC1, CDK1, TACC3, MCM7, EXO1, TPX2, BUB1, ANLN, ESPL1, KPNA2, AURKB, FEN1, NUSAP1, CCNB2, HMMR, CKAP2, INCENP, MKI67, BIRC5, HELLS, ZWILCH, TOP2A, ERCC6L and INCENP were the hub genes (Figure S1).
Characteristics of prognostic DEIGs
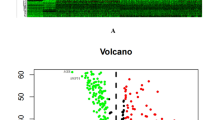
As expected, the inflammatory pathways were indicated as the most frequently implicated by gene functional enrichment analysis. Regulation of leukocyte activation, extracellular matrix and cell adhesion molecule binding were the most frequent GO terms (Fig. 1a). The cytokine−cytokine receptor interaction was the top term enriched by differentially expressed prognostic DEIGs (Fig. 1b). We also found that the missense is the most common type of mutations by examining genetic alterations of these genes (Figure S2).

Gene functional enrichment of differentially expressed immune-related genes (aTop10 enriched KEGG gene sets; b GO analyses of the prognostic DEGs in the categories of biological processes (BP), cellular components (CC), and molecular functions (MF))
Prognostic modeling, identification of an optimal prognostic signature using immune related genes
The prediction model was built on the most frequent gene set with effective coefficients in the lasso regression. Model 1, Model 2, Model 3 and Model 4 were respectively constructed using the top 100, 159, 200 and 436 DEIGs. We found that model 4 which was correlated with tumor burden, tumor stage and metastasis, performed best in prognostic predictions. The optimized model consists of the following genes: CAMP, CCT6A, CDH17, EFNB2, FKBP3, GATA2, ITIH4, SMAD9, P2RX1, PFKP, PKP2, PTGFRN, PTPRH, CCL20, SSR4, KLF10, UPK1B, SLC7A5, FKBP6, FERMT2, FLRT1, DDIT4, LY6K, NLRP2, HAPLN2, CCNL2, EMR3, COL27A1, TSLP, SFXN1, WFIKKN2, PCSK9, IZUMO1. The list of coefficients for those genes are shown in Supplementary Information (Table S2, Figure S3). The ROC curve was 0.824 for 3 years, 0.838 for 5 years, 0.834 for 10 years, indicating the prognostic model based on DEIGs has definite potential in survival monitoring (Figs. 2 and 3, S4). Univariate Cox regression analysis suggested that the prognostic signature, age, tumor stage, pathologic stage and metastasis status are all associated with prognosis (Table 1). The prognostic model based on DEIGs was identified as an independent predictor by using multivariate cox regression analysis after the adjustment of other parameters (Fig. 4).

ROC curve validation of prognostic value of the prognostic index of each model (a Modle-1, Input gene list: top100 sorted immune-related genes by p value; b Modle-2, Input gene list: top159 sorted immune-related genes by p value; c Modle-3, Input gene list: top200 sorted immune-related genes by p value; d Modle-4, Input gene list: top436sorted immune-related genes by p value)

Identification of an immune signature predicting prognosis risk of patients in LUAD using model 4 (a survival analysis of the training dataset; b survival analysis in the testing data; c The heatmaps distinct gene expression profiles of the cases belonging to the high and low risk score groups)

Multivariate cox regression analysis
Correlation between prognostic signature and immune infiltration
We analyzed the relationship between model predicted risk score and immune cell infiltration to see if the DEIGs accurately reflected the status of tumor immune microenvironment. The risk score of our model is inversely related to the abundances of infiltrated immune cells as well as classical markers for immune cells, including CD8+ T cell, CD4+ T cell, B cell and dendritic cell (Fig. 5, S5, S6, S7 and S8).

Relationships between the risk score and estimated infiltration abundances of immune cells
The relationship between the expression of FERMT2, FKBP3, SMAD9, GATA2, IHIH4 and the overall survival of LUAD
In order to verify the clinical value of the model, we finally examined the expression of FERMT2, FKBP3, SMAD9, GATA2 and ITIH4 in 30 lung adenocarcinoma tissues by immunohistochemistry, considering the availability of antibodies. 86.67% (26/30) of LUAD patients tissue samples had positive expression of FERMT2, 83.33% (25/30) of FKBP3, 26.67% (8/30) of SMAD9, 23.33% (7/30) of GATA2 and 20.00% (6/30) of ITIH4 (Fig. 6). Based on the result of IHC of FERMT2, FKBP3, SMAD9, GATA2 and ITIH4, we divided the patients into 2 groups (negative group and positive group); the characteristics of each group are shown in Table 2.We found that the positive expression of FERMT2, FKBP3, SMAD9, GATA2 and ITIH4 had a correlation with the TNM stage, cellular differentiation and the lymph node metastasis (p < 0.05). No significant correlation was found with the age and sex (p > 0.05). We found that 83.33 and 91.67% of LUAD patients tissues in stage I-II (15/18) and stage III-IV (11/12) had positive expression of FERMT2(P < 0.05). 83.33% of LUAD patients tissues in stage I-II (15/18) and stage III-IV (10/12) had positive expression of FKBP3 (P > 0.05). Meanwhile, the positive rate of SMAD9, GATA2 and ITIH4 were 27.78% (5/18),16.67% (3/18) and 1.11% (2/18) in stage I-II and 25.00%(3/12),33.33%(4/12) and 33.33% (4/12) in stage III-IV(P < 0. 05). These were consistent with the results of our survival analysis: high levels of FERMT2, FKBP3 and low levels of SMAD9, ITIH4, GATA2 expression are associated with poor overall survival in LUAD.

Immunohistochemical staining of FERMT2, FKBP3, SMAD9, GATA2 and IHITH4 protein in LUAD tissues (magnification, × 200). A1:weak expression of FERMT2;A2:moderate expression of FERMT2;A3:strong expression of FERMT2; B1:weak expression of FKBP3; B2:moderate expression of FKBP3;B3:strong expression of FKBP3;C1:weak expression of GATA2;C2:moderate expression of GATA2; C3:strong expression of GATA2;D1weak expression of IHITH4;D2:moderate expression of IHITH4;D3:strong expression of IHITH4; E1:weak expression of SMAD9;E2:moderate expression of SMAD9;E3:strong expression of SMAD9
Then Kaplan–Meier was performed to determine the effect of the immune related genes on prognosis of LUAD patients. Univariate Cox regression analysis demonstrated that the expression of FERMT2(HR = 5.084, 95% CI, 2.569 ~ 8.215), FKBP3(HR = 3.186, 95% CI, 2.279 ~ 7.945), SMAD9(HR = 0.791, 95%CI = 0.769 ~ 0.913), GATA2 (HR = 0.801, 95%CI = 0.744 ~ 0.952) and ITIH4 (HR = 0.776, 95%CI = 0.695 ~ 0.889) were significantly associated with overall survival (OS) (Fig. 7). Of note, The detailed coefficients of these five genes are 0.242851(FERMT2), 0.168033(FKBP3), − 0.00976(SMAD9), − 0.04737(GATA2) and − 0.0019 (ITIH4). The signs of those coefficients are consistent with the roles of the expression of those genes as revealed by survival analysis.

Effect of FERMT2, FKBP3, SMAD9, GATA2 and IHITH4 status on OS of LUAD patients. (a: Median survival time (MS) of LUAD patients with FERMT2 (−) and FERMT2 (+) was 41.4 months (95% CI, 37.1 to 45.2) and 21.70(95% CI,17.2 to 25.16), respectively, P = 0.0241. b: The MS of patients with FKBP3 (−) and FKBP3 (+) was 41.15 months (95% CI,36.23 to 46.72) and 20.25 (95% CI, 16.79 to 23.15), respectively, P = 0.0044. c: The MS of patients with SMAD9 (−) and SMAD9 (+) was 18.75 months (95% CI,13.15 to 23.38) and 34.05 (95% CI, 29.12 to 38.87), respectively, P = 0.0030. d: The MS of patients with GATA2 (−) and GATA2 (+) was 19.00 months (95% CI,14.47 to 24.13) and 37.10 (95% CI, 32.42 to 42.51), respectively, P = 0.0123. e: The MS of patients with ITIH4 (−) and ITIH4 (+) was 20.25 months (95% CI,15.14 to 25.42) and 34.05 (95% CI, 30.18 to 41.92), respectively, P = 0.0165)
Discussion
Adenocarcinoma is the most common pathological type of lung cancer with highly invasive and fatal. Most patients’ overall survival is less than 5 years whom were diagnosed at advanced stage [26]. Existing treatments extend the survival of part of patients with lung adenocarcinoma, but the overall curative effect is not so good, especially in the advanced cases [27, 28]. The shortage of effective prognostic biomarkers to guide therapy is one of the reasons for the poor prognosis [29]. Therefore, there is a need to construct an efficient prognostic model to develop individualized treatment plans for patients and improve the prognosis of LUAD.
Current studies have found that the development of cancer are not only dependent on tumor cell characteristics but are also affected by the interaction with infiltrated immunocytes [30, 31]. The tumors with higher immune cells and mediators proportion were proved to be more effective to the immune treatment [32]. There is mounting evidence supporting that the immunogenomics and immune microenvironment play an important role in cancer [33, 34]. As an example, at the levels of DNA, RNA and the epigenome, Rosenthal et al. has observed the signs of immunologic sculpting, immunoediting, and immune escape [35]. These studies provide the clues for our research toward DEIGs. In our study, the DEIGs were identified by the bioinformatics analysis with TCGA datasets, we found that the inflammatory pathway was an inseparable aspect of tumor development. Similar results were found in other studies [36,37,38].
Four prediction models were built with lasso regression using distinct lists of immune related genes. Model 4 which contains 33 prognosis DEIGs performed best in prognostic predictions, and correlated with tumor burden, tumor stage and metastasis. Among those prognosis-specific immune related genes, 14(e.g., CCT6A, EFNB2, FKBP3, FERMT2, SMAD9, GATA2, PFKP, PKP2, PTPRH, CCL20, SLC7A5, DDIT4, LY6K, ITIH4) have been demonstrated to be participate in the the pathogenesis of cancer or reported to be significant predictors of survival [39,40,41,42,43,44,45,46]. This implies that our analysis has certain theoretic value. The remaining genes which have not been reported could serve as new potential biomarkers of LUAD.
On one hand, the coef of FERMT2 and FKBP3 were highest, and the expression of FERMT2, FKBP3, SMAD9, GATA2 and ITIH4 in the tissues of LUAD patients and their correlation with patient survival have not been studied. On the other hand, considering the availability of antibodies, we finally examined the expression of FERMT2, FKBP3, SMAD9, GATA2 and ITIH4 in 30 LUAD tissues by immunohistochemistry. Previous studies showed that FERMT2 highly expressed in NSCLC, esophageal squamous cancer, breast cancer, cholangiocarcinoma and pancreatic cancer, and can affect the migration ability of tumor cells and disease progression [47,48,49]. Guo et al. found the expression of FERMT2 is closely correlated with the tumor clinical stage of lung cancer [50]. Our findings concordant with these results. It is hypothesized that FERMT2 may have effects on tumor immunity through interactions with integrin-like protein. A large number of studies have proved that HDACs are involved in regulating the innate and adaptive immune processes of the body [51]. FKBP3 which is a member of FK506-binding proteins, could promote proliferation of lung cancer cells through regulating Sp1/HDAC2/p27 [52], we assumed that its immunoregulation effects could be related to HDAC2 [53]. Meanwhile, there is plenty of evidence that SMAD9, ITIH4 and GATA2 have close connection with the initiation, progression and prognosis of various malignancies including lung cancer [54,55,56,57]. SMAD9 is located on chromosome 13q13.3 and encodes a protein that is a member of the SMAD family, which is a crucial pathway for the TGF-β transcription factor family [58]. It was found that SMAD9 may be regulated by methylation, phosphorylation and dephosphorylation in the occurrence and development of lung cancer [59]. Previous studies suggested that GATA2 is important for survival and growth of NSCLC cells with mutations in KRAS and other oncogenes on the RTK/RAS pathway. The deletion of GATA2 reduces survival of KRAS mutant NSCLC cells significantly inhibit the development of NSCLC [60]. In addition, recent study reported that GATA2 is sufficient to drive PD-L1 and PD-L2 expression and is necessary for PD-L2 expression. It was reported that cytokines, such as IL-6, TNF-α, IL-10 and lipopolysaccharide (LPS) influence the expression of ITIH4. ITIH4, as an inflammation biomarker may participate in immune regulation through JAK/STAT [61].
In our study, we also revealed that the expression of FERMT2, FKBP3, SMAD9, ITIH4 and GATA2 are independent prognostic factors, furthermore, high levels of FERMT2, FKBP3 and low levels of SMAD9, ITIH4, GATA2 expression are associated with poor overall survival in LUAD.
Combined with the TCGA database analysis and literature reports in this study, we speculated that the expression of these genes that influence tumor prognosis are significantly correlated with multiple cytokine pathways and immunity correlation reaction.
As we know, the tumor immune microenvironment was composed of various infiltrating immune cells including T cells, B cells, natural killer cells, dendritic cells, myeloid-derived suppressor cells, neutrophils, and macrophages [62, 63]. Lots of studies have demonstrated the relationship between the tumor-infiltrating immune cells and tumor growth, metastasis or angiogenesis of lung cancer [64,65,66]. These reports are in line with our results. We found that the risk score of our model was inversely related to the infiltration of various immune cells, as well as the markers of B cell, CD4+ T cell, CD8+ T cell and dendritic cell. These results indicated that the high-risk patients’ infiltration levels of immune cells might be lower, suggesting that the abnormal expression of immune genes can lead to the disorder of tumor immune microenvironment, and then participate in the occurrence, development, invasion and metastasis of LUAD.
Conclusions
In this study, we constructed 4 models to predict the prognosis of patients with LUAD, and proposed an optimal prognostic model, our preliminary results hint at the correlation between immune related genes and the prognosis of LUAD. However, the further research about the mechanisms of the DEIGs modulate the progression of LUAD is needed.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Supplementary Information files. The datasets generated and/or analyzed during the current study are available in the TCGA repository (https://portal.gdc.cancer.gov/). The immune-related genes were derived from InnateDB (https://www.innatedb.com/). While the estimated infiltration abundance of immune cells of LUAD samples were obtained by TIMER (https://cistrome.shinyapps.io/timer/).
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- NSCLC:
-
Non small-cell lung cancer
- LUAD:
-
Lung adenocarcinoma
- DEIGs:
-
Differentially expressed immune genes
- TME:
-
Tumor microenvironment
- TMB:
-
Tumor mutational burden
- MMR:
-
Mismatch repair
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein-protein interaction
- ROC:
-
Receiver operating characteristic curve
- IHC:
-
Immunohistochemistry
- HRP:
-
Horseradish peroxidase
- IRS:
-
Immunoreactivity score
References
Global Burden of Disease Cancer C, Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, Abdel-Rahman O, Abdelalim A, Abdoli A, Abdollahpour I, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol. 2019;5(12):1749–68.
Cacho-Diaz B, Garcia-Botello DR, Wegman-Ostrosky T, Reyes-Soto G, Ortiz-Sanchez E, Herrera-Montalvo LA. Tumor microenvironment differences between primary tumor and brain metastases. J Transl Med. 2020;18(1):1.
Ling B, Huang Z, Huang S, Qian L, Li G, Tang Q. Microenvironment analysis of prognosis and molecular signature of immune-related genes in lung adenocarcinoma. Oncol Res. 2020. https://doi.org/10.3727/096504020X15907428281601.
Santaniello A, Napolitano F, Servetto A, De Placido P, Silvestris N, Bianco C, Formisano L, Bianco R. Tumour microenvironment and immune evasion in EGFR addicted NSCLC: hurdles and possibilities. Cancers (Basel). 2019;11(10):1419.
Qi X, Qi C, Qin B, Kang X, Hu Y, Han W. Immune-stromal score signature: novel prognostic tool of the tumor microenvironment in lung adenocarcinoma. Front Oncol. 2020;10:541330.
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830.e814.
Saab S, Zalzale H, Rahal Z, Khalifeh Y, Sinjab A, Kadara H. Insights into lung cancer immune-based biology, prevention, and treatment. Front Immunol. 2020;11:159.
Chen X, Zhang Z, Hou X, Zhang Y, Zhou T, Liu J, Lin Z, Fang W, Yang Y, Ma Y, et al. Immune-related pneumonitis associated with immune checkpoint inhibitors in lung cancer: a network meta-analysis. J Immunother Cancer. 2020;8(2):e001170.
Kobayashi H. Efficacy and tolerance of immune checkpoint inhibitors for non-small cell lung cancer patients with interstitial lung disease - Reply. Respirology. 2020;25(8):892-3.
Paulsen EE, Kilvaer TK, Khanehkenari MR, Al-Saad S, Hald SM, Andersen S, Richardsen E, Ness N, Busund LT, Bremnes RM, et al. Assessing PDL-1 and PD-1 in non-small cell lung cancer: a novel Immunoscore approach. Clin Lung Cancer. 2017;18(2):220–233.e228.
Wagener-Ryczek S, Buettner R. The value of tumor mutational burden to select patients for immunotherapy. Expert Rev Anticancer Ther. 2021;21(1):1-3.
Wagle MC, Castillo J, Srinivasan S, Holcomb T, Yuen KC, Kadel EE, Mariathasan S, Halligan DL, Carr AR, Bylesjo M, et al. Tumor fusion burden as a hallmark of immune infiltration in prostate cancer. Cancer Immunol Res. 2020;8(7):844–50.
Cedres S, Ponce-Aix S, Iranzo P, Callejo A, Pardo N, Navarro A, Martinez-Marti A, Gomez-Abecia S, Zucchiatti AC, Sansano I, et al. Analysis of mismatch repair (MMR) proteins expression in a series of malignant pleural mesothelioma (MPM) patients. Clin Transl Oncol. 2020;22(8):1390–8.
Evans M, O'Sullivan B, Smith M, Taniere P. Predictive markers for anti-PD-1/PD-L1 therapy in non-small cell lung cancer-where are we? Transl Lung Cancer Res. 2018;7(6):682–90.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71.
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–33.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–10.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22.
Fung AS, Leighl NB. Advancing the systemic therapy of lung cancer: quality or quantity? Lancet Oncol. 2020;21(3):322–4.
Sethi RS, Brar RS, Singh O, Singh B. Immunolocalization of pulmonary intravascular macrophages, TLR4, TLR9 and IL-8 in normal and Pasteurella multocida-infected lungs of water buffalo (Bubalus bubalis). J Comp Pathol. 2011;144(2–3):135–44.
Xu Z, Wu Z, Zhang J, Zhou R, Ye L, Yang P, Yu B. Development and validation of an oxidative phosphorylation-related gene signature in lung adenocarcinoma. Epigenomics. 2020;12(15):1333–48.
Alhejaili F, Al-Hajeili M, Bakhsh SI, Banjar NS, Alghamdi W, Alsulami AF, Algamdi M. Survival of patients with small cell lung cancer in King Abdulaziz University Hospital, Jeddah, Saudi Arabia. Cureus. 2020;12(1):e6648.
Fukui T, Hosotani S, Soda I, Ozawa T, Kusuhara S, Kakegawa MI, Kasajima M, Hiyoshi Y, Igawa S, Yokoba M, et al. Current status and progress of concurrent chemoradiotherapy in patients with locally advanced non-small cell lung cancer prior to the approval of durvalumab. Thorac Cancer. 2020;11(4):1005–14.
Zong Q, Zhu F, Wu S, Peng L, Mou Y, Miao K, Wang Q, Zhao J, Xu Y, Zhou M. Advanced pneumonic type of lung adenocarcinoma: survival predictors and treatment efficacy of the tumor. Tumori. 2020:300891620947159. https://doi.org/10.1177/0300891620947159.
Qin FL, Xu ZY, Yuan LQ, Chen WJ, Wei JB, Sun Y, Li SK. Novel immune subtypes of lung adenocarcinoma identified through bioinformatic analysis. FEBS Open Bio. 2020;10(9):1921–33.
Dora D, Rivard C, Yu H, Bunn P, Suda K, Ren S, Lueke Pickard S, Laszlo V, Harko T, Megyesfalvi Z, et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol Oncol. 2020;14(9):1947–65.
Domagala-Kulawik J. New frontiers for molecular pathology. Front Med (Lausanne). 2019;6:284.
Li J, Li X, Zhang C, Zhang C, Wang H. A signature of tumor immune microenvironment genes associated with the prognosis of nonsmall cell lung cancer. Oncol Rep. 2020;43(3):795–806.
Kudo Y, Haymaker C, Zhang J, Reuben A, Duose DY, Fujimoto J, Roy-Chowdhuri S, Solis Soto LM, Dejima H, Parra ER, et al. Suppressed immune microenvironment and repertoire in brain metastases from patients with resected non-small-cell lung cancer. Ann Oncol. 2019;30(9):1521–30.
Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, Lund T, Tanic M, Reading JL, Joshi K, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567(7749):479–85.
Wang X, Yin H, Zhang L, Zheng D, Yang Y, Zhang J, Jiang H, Ling X, Xin Y, Liang H, et al. The construction and analysis of the aberrant lncRNA-miRNA-mRNA network in non-small cell lung cancer. J Thorac Dis. 2019;11(5):1772–8.
Kobayashi S, Karube Y, Matsumura Y, Nishihira M, Inoue T, Araki O, Maeda S, Chida M. Inflammatory risk factors for early recurrence of non-small cell lung cancer within one year following curative resection. World J Surg. 2020;44(10):3510–21.
Narayanapillai SC, Han YH, Song JM, Kebede ME, Upadhyaya P, Kassie F: Modulation of the PD-1/PD-L1 immune checkpoint axis during inflammation-associated lung tumorigenesis. Carcinogenesis. 2020;41(11):1518-28.
Zeng G, Wang J, Huang Y, Lian Y, Chen D, Wei H, Lin C, Huang Y. Overexpressing CCT6A contributes to cancer cell growth by affecting the G1-to-S phase transition and predicts a negative prognosis in hepatocellular carcinoma. Onco Targets Ther. 2019;12:10427–39.
Ni Q, Chen P, Zhu B, Li J, Xie D, Ma X. Expression levels of EPHB4, EFNB2 and caspase-8 are associated with clinicopathological features and progression of esophageal squamous cell cancer. Oncol Lett. 2020;19(1):917–29.
Chen G, Liu H, Zhang Y, Liang J, Zhu Y, Zhang M, Yu D, Wang C, Hou J. Silencing PFKP inhibits starvation-induced autophagy, glycolysis, and epithelial mesenchymal transition in oral squamous cell carcinoma. Exp Cell Res. 2018;370(1):46–57.
Sato T, Soejima K, Arai E, Hamamoto J, Yasuda H, Arai D, Ishioka K, Ohgino K, Naoki K, Kohno T, et al. Prognostic implication of PTPRH hypomethylation in non-small cell lung cancer. Oncol Rep. 2015;34(3):1137–45.
Song Q, Shang J, Zhang C, Chen J, Zhang L, Wu X. Transcription factor RUNX3 promotes CD8(+) T cell recruitment by CCL3 and CCL20 in lung adenocarcinoma immune microenvironment. J Cell Biochem. 2020;121(5–6):3208–20.
Hao XL, Tian Z, Han F, Chen JP, Gao LY, Liu JY. Plakophilin-2 accelerates cell proliferation and migration through activating EGFR signaling in lung adenocarcinoma. Pathol Res Pract. 2019;215(7):152438.
Alfarsi LH, El-Ansari R, Craze ML, Masisi BK, Mohammed OJ, Ellis IO, Rakha EA, Green AR. Co-expression effect of SLC7A5/SLC3A2 to predict response to endocrine therapy in oestrogen-receptor-positive breast cancer. Int J Mol Sci. 2020;21(4):1407.
Benti S, Tiwari PB, Goodlett DW, Daneshian L, Kern GB, Smith MD, Uren A, Chruszcz M, Shimizu LS, Upadhyay G. Small molecule binds with lymphocyte antigen 6K to induce cancer cell death. Cancers (Basel). 2020;12(2):509.
Guo L, Wu C. Mechano-regulation of proline metabolism and cancer progression by kindlin-2. Mol Cell Oncol. 2019;6(3):1596003.
Sossey-Alaoui K, Pluskota E, Bialkowska K, Szpak D, Parker Y, Morrison CD, Lindner DJ, Schiemann WP, Plow EF. Kindlin-2 regulates the growth of breast cancer tumors by activating CSF-1-mediated macrophage infiltration. Cancer Res. 2017;77(18):5129–41.
Yoshida N, Masamune A, Hamada S, Kikuta K, Takikawa T, Motoi F, Unno M, Shimosegawa T. Kindlin-2 in pancreatic stellate cells promotes the progression of pancreatic cancer. Cancer Lett. 2017;390:103–14.
Guo WH, Bian JJ, Tian GF, Lyu ZX, Gui YX, Ye L. Expression of Fermintin family homologous protein 2 in non-small cell lung cancer and its clinical significance. Zhonghua Bing Li Xue Za Zhi. 2018;47(10):780–3.
Hadley M, Noonepalle S, Banik D, Villagra A. Functional analysis of HDACs in tumorigenesis. Methods Mol Biol. 1983;2019:279–307.
Zhu W, Li Z, Xiong L, Yu X, Chen X, Lin Q. FKBP3 promotes proliferation of non-small cell lung cancer cells through regulating Sp1/HDAC2/p27. Theranostics. 2017;7(12):3078–89.
Tong J, Shen Y, Chen X, Wang R, Hu Y, Zhang X, Zhang Z, Han L. FKBP3 mediates oxaliplatin resistance in colorectal cancer cells by regulating HDAC2 expression. Oncol Rep. 2019;42(4):1404–12.
Kokaji E, Shimomura A, Minamisaka T, Nakajima T, Miwa S, Hatta H, Nishida T, Kiya C, Imura J. Endoglin (CD105) and SMAD4 regulate spheroid formation and the suppression of the invasive ability of human pancreatic cancer cells. Int J Oncol. 2018;52(3):892–900.
Yang J, Xiong X, Liu S, Zhu J, Luo M, Liu L, Zhao L, Qin Y, Song T, Huang C. Identification of novel serum peptides biomarkers for female breast cancer patients in Western China. Proteomics. 2016;16(6):925–34.
Woo J, Santasusagna S, Banks J, Pastor-Lopez S, Yadav K, Carceles-Cordon M, Dominguez-Andres A, Den RB, Languino LR, Pippa R, et al. Urine extracellular vesicle GATA2 mRNA discriminates biopsy result in men with suspicion of prostate cancer. J Urol. 2020;204(4):691–700.
Zhang L, Gao L, Shao M, Sun GY. A MYC target long non-coding RNA GATA2-AS1 regulates non-small cell lung cancer growth. Neoplasma. 2019;66(6):954–62.
Dexheimer V, Gabler J, Bomans K, Sims T, Omlor G, Richter W. Differential expression of TGF-beta superfamily members and role of Smad1/5/9-signalling in chondral versus endochondral chondrocyte differentiation. Sci Rep. 2016;6:36655.
Gao L, Tian Q, Wu T, Shi S, Yin X, Liu L, Zheng L, Wang P, Tian Y, Xu S. Reduction of miR-744 delivered by NSCLC cell-derived extracellular vesicles upregulates SUV39H1 to promote NSCLC progression via activation of the Smad9/BMP9 axis. J Transl Med. 2021;19(1):37.
Tessema M, Yingling CM, Snider AM, Do K, Juri DE, Picchi MA, Zhang X, Liu Y, Leng S, Tellez CS, et al. GATA2 is epigenetically repressed in human and mouse lung tumors and is not requisite for survival of KRAS mutant lung cancer. J Thorac Oncol. 2014;9(6):784–93.
Ma Y, Li R, Wang J, Jiang W, Yuan X, Cui J, Wang C. ITIH4, as an inflammation biomarker, mainly increases in bacterial bloodstream infection. Cytokine. 2021;138:155377.
Hu ZQ, Xin HY, Luo CB, Li J, Zhou ZJ, Zou JX, Zhou SL. Associations among the mutational landscape, immune microenvironment, and prognosis in Chinese patients with hepatocellular carcinoma. Cancer Immunol Immunother. 2020. https://doi.org/10.1007/s00262-020-02685-7.
Stankovic B, Aamodt H, Bjorhovde HAK, Muller E, Hammarstrom C, Brustugun OT, Helland A, Oynebraten I, Corthay A. The immune microenvironment in typical carcinoid lung tumour, a brief report of four cases. Scand J Immunol. 2020;92(2):e12893.
Amrein MA, Buhrer ED, Amrein ML, Li Q, Rothschild S, Riether C, Jaggi R, Savic-Prince S, Bubendorf L, Gautschi O, et al. Chemotherapy negatively impacts the tumor immune microenvironment in NSCLC: an analysis of pre- and post-treatment biopsies in the multi-center SAKK19/09 study. Cancer Immunol Immunother. 2020. https://doi.org/10.1007/s00262-020-02688-4.
Tamminga M, Hiltermann TJN, Schuuring E, Timens W, Fehrmann RS, Groen HJ. Immune microenvironment composition in non-small cell lung cancer and its association with survival. Clin Transl Immunol. 2020;9(6):e1142.
Boys J, Onaitis M. Commentary: breaking down non-small cell lung cancer tumor microenvironment heterogeneity and predicting response to immune checkpoint inhibitors. J Thorac Cardiovasc Surg. 2020;159(4):1612–3.
Acknowledgements
We thank Miaomiao Jiang and Xiaoyun Huang from Zhiyu Center for Systems Biology (Intelliphecy Inc.) for support with Bioinformatics.
Funding
This work was supported by National Natural Science Foundation of Shaanxi (No. 2019JQ-566).
Author information
Authors and Affiliations
Contributions
Yang Zhai and Qian Chen contributed to the conception of the study. Bin Zhao and Yuzhen Wang contributed significantly to analysis and manuscript preparation; Lina Li, Jingjin Li, Xu Li and Linhan Chang performed the data analyses and wrote the manuscript; Qian Chen, Zijun Liao helped perform the analysis with constructive discussions. Yang Zhai and Qian Chen wrote the main manuscript text and Bin Zhao and Yuzhen Wang prepared figures 1-3. Lina Li 1and Jingjin Li prepared figures 4-5. Xu Li and Linhan Chang prepared figures 6-7. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by Ethics Committee of Tumor Hospital of Shaanxi Province. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Consent for publication
No consent is required in this study.
Competing interests
These authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
A total of 436 differentially expressed genes were identified. Figure S1. PPI network inferred by STRING using 436 differentially expressed immune genes with p.value < 0.05 in cox test. Hub genes were identified by cytoscape. Shown is cluster1 with 31 genes. Figure S2. Mutation landscape of 436 target genes (top50) in 569 tumors. Figure S3. The coefficients for the genes retained in model 4. Table S2. The list of coefficients for genes retained in model 4. Figure S4. Identification of an immune signature predicting prognosis risk of patients in LUAD using each models (A1,B1,C1: A cutoff of risk factor for modle 1, modle 2, modle 3; A2,B2,C2: Survival analysis of the training dataset for modle 1, modle 2, modle 3; A3,B3,C3: Survival analysis in the testing data for modle 1, modle 2, modle 3). Figure S5. Relationships between the risk score and CD8+ T cell markers. Figure S6. Relationships between the risk score and CD4+ T cell markers. Figure S7. Relationships between the risk score and B cell markers. Figure S8. Relationships between the risk score and dendritic cell markers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhai, Y., Zhao, B., Wang, Y. et al. Construction of the optimization prognostic model based on differentially expressed immune genes of lung adenocarcinoma. BMC Cancer 21, 213 (2021). https://doi.org/10.1186/s12885-021-07911-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-07911-8