
Abstract
Background
Acute lymphoblastic leukemia (ALL), the most common childhood malignancy, is characterized by recurring structural chromosomal alterations and genetic alterations, whose detection is critical in diagnosis, risk stratification and prognostication. However, the genetic mechanisms that give rise to ALL remain poorly understood.
Methods
Using next-generation sequencing (NGS) in matched germline and tumor samples from 140 pediatric Chinese patients with ALL, we landscaped the gene mutations and estimated the mutation frequencies in this disease.
Results
Our results showed that the top driver oncogenes having a mutation prevalence over 5% in childhood ALL included KRAS (8.76%), NRAS (6.4%), FLT3 (5.7%) and KMT2D (5.0%). While the most frequently mutated genes were KRAS, NRAS and FLT3 in B cell ALL (B-ALL), the most common mutations were enriched in NOTCH1 (23.1%), FBXW7 (23.1%) and PHF6 (11.5%) in T cell ALL (T-ALL). These mutant genes are involved in key molecular processes, including the Ras pathway, the Notch pathway, epigenetic modification, and cell-cycle regulation. Strikingly, more than 50% of mutations occurred in the high-hyperdiploid (HeH) ALL existed in Ras pathway, especially FLT3 (20%). We also found that the epigenetic regulator gene KMT2D, which is frequently mutated in ALL, may be involved in driving leukemia transformation, as evidenced by an in vitro functional assay.
Conclusion
Overall, this study provides further insights into the genetic basis of ALL and shows that Ras mutations are predominant in childhood ALL, especially in the high-hyperdiploid subtype in our research.
Similar content being viewed by others


Background
Acute lymphoblastic leukemia (ALL), the most common childhood tumor, results from the clonal proliferation of lymphoid stem or progenitor cells with arrested maturation, with more than 80% originating from B cell progenitors [1]. ALL is characterized by recurring structural chromosomal alterations, including aneuploidy (high-hyperdiploid, chromosomes ≥51; hypodiploid, chromosomes ≤44) and translocations (e.g., t (12;21)/ETV6-RUNX1, t (1;19)/TCF3-PBX1, t (9;22)/BCR-ABL1, and KMT2A (also known as MLL) rearrangement). However, chromosomal changes alone are often insufficient to trigger leukemia, some additional genetic alterations must contribute to tumorigenesis [2, 3].
Cytogenetic alterations or molecular abnormalities are frequent, and several molecular markers have been identified to stratify risk and predict prognosis, as they play key roles in ALL pathogenesis. Specific ALL subtypes exhibit different mutation distributions; for example, TP53 mutations mostly occur in hypodiploidy [4, 5]. PAX5/IKZF1 copy number abnormalities frequently exist in B-ALL, whereas mutations within NOTCH1, FBXW7, and CDKN2A/CDKN2B are enriched in T-ALL [1, 6,7,8]. Rare germline mutations in the genes PAX5 [9] and ETV6 [10] were found to be linked to familial leukemia, and some chemical agents or radiation exposure could increase the incidence of leukemia [6]. In addition, some molecular alterations, such as CREBBP [11,12,13], NT5C2 [14, 15] and PRPS1 mutations [16], are associated with chemo-resistance. Thus, the identification of these abnormalities not only reveals molecular pathology, but also provides important therapeutic targets. Some targetable alterations or pathways have been used for therapeutic interventions in the clinic, especially kinase-activating alterations in BCR-ABL1-positive or Philadelphia chromosome-like ALL patients who are amenable to tyrosine kinase inhibitors with improved survival rates [17, 18]. However, a substantial percentage of patients classified as having a “good prognosis” (e.g., t (12;21)/ETV6-RUNX1 or high-hyperdiploid) still experience relapse, which may be caused by the existence of additional or secondary molecular variants. Therefore, it remains important to further identify the repertoires of gene mutations and understand its clinical significance in pediatric ALL.
Recently, genetic profiling of several subtypes of pediatric ALL has been conducted with NGS [4, 5, 11, 19, 20]. Numerous germline genetic variants and somatic alterations have been identified in newly diagnosed and relapsed childhood ALL or in specific subtypes, which may also have prognostic implications [19, 20]. NGS has revealed changes in the microarchitecture and gene sequence, which advanced the understanding of the molecular basis of ALL and complemented genetic features of the ALL subtypes.
In this study, we used targeted exome sequencing technology to reveal the mutational spectrum in patients with ALL at initial diagnosis to better understand the cytogenetic and molecular classification of pediatric ALL in Chinese children, which may lead to the discovery of new therapeutic targets and enable the development of a tailored therapeutic regimen for each patient.
Methods
Sample collection and genomic DNA extraction
A total of 140 pediatric patients (≤18 years) with ALL enrolled consecutively in this study were newly diagnosed and treated in the children’s hospital of Fudan University in China between January 2015 and December 2017. ALL diagnosis was established by analysis of leukemic cells with morphology, immunophenotyped, and cytogenetics. Immunophenotype (B-ALL or T-ALL) was defined according to the European Group for the Immunological Characterization of Leukemias. Informed consent was obtained in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Fudan Institutes. Bone marrow samples were collected at initial diagnosis; matched remission samples or fingernails were used as germline controls. Genomic DNA was extracted from cell pellets using DNAeasy Blood and Tissue Kit (Qiagen, USA). DNA was quantified using a Qubit Fluorometer (Life Technologies, USA), and DNA integrity was assessed by agarose gel electrophoresis. The transcripts of BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, SIL-TAL1 fusion genes, and MLL rearrangement (MLLr) were detected with reverse transcriptase polymerase chain reaction (RT-PCR) or fluorescence in situ hybridization (FISH) as previously described [21].
Targeted capture sequencing and mutation analysis
Targeted capture libraries were prepared, and the exons of 950 genes related to cancer were selected for sequencing (Table S1). Genomic DNA samples were sheared by sonication, and the sheared genomic DNA was then hybridized with a NimbleGen 2.0 probe sequence capture array of Roche (http://www.nimblegen.com/products/seqcap/ez/v2/index.html) to enrich the exonic DNA (Joy Orient, China). The libraries were first tested for enrichment by qPCR and for size distribution and concentration using the Agilent Bioanalyzer 2100. The samples were then sequenced on an Illumina Hiseq2500, and two parallel reactions were performed for each sample. Raw image files were processed by BclToFastq (Illumina) for base calling and generating the raw data. The low-quality variations were filtered out using a quality score ≥ 20 (Q20). The sequencing reads were aligned to the NCBI human reference genome (hg19) using BWA (version 0.5.10), including coverage and quality assessment, single-nucleotide variant (SNV) and indel detection, annotation and prediction of deleterious effects for sequence mutations. Samtools and Pindel were used to analyze single-nucleotide polymorphisms (SNPs) and indels in the sequence. Synonymous changes and SNPs with MAF (minor allele frequency) higher than 5% were removed (http://www.ncbi.nlm.nih.gov/projects/SNP). Nonsynonymous changes and small indels were filtered using SIFT (version 1.03), PolyPhen-2 (version 2.2.2), PROVEAN (version 1.1.3), and MutationTaster 2. All candidate mutations were filtered with minimum coverage ≥10, minimum tumor variant frequency ≥ 0.10, normal variant frequency ≤ 0.05, and candidate driver mutations were considered as two prediction algorithms to be significant or identified as recurrent in COSMIC.
Generation of KMT2D knockdown cell lines
Lentivirus-mediated gene-specific small hairpin RNAs (shRNAs) were used to knockdown the expression of the KMT2D gene in Nalm-6 cells (human ALL pre-B cells). Nalm-6 cells were obtained from FuDan IBS Cell Center (FDCC) and tested for mycoplasma (catalog no. FDCC-HGN101) and were cultured in RPMI1640 (Gibco, USA) medium with 10% FBS (Gibco, USA) and 1% penicillin-streptomycin (Gibco, USA). shRNA-targeted sequences (Table S2) were subcloned into the lentiviral vector pLenO-GTP, and then plasmids and packaging vectors (pRsv-REV, pMDlg-pRRE, pMD2G, pLenO-GTP) were cotransfected into HEK293T cells to generate lentivirus. These vectors were obtained from BioLink Laboratory (Shanghai, China). A total of 5 × 104 cells/μl were infected with MOI = 100 IU/ml virus and 5 μg/ml of polybrene (Sigma-Aldrich, Germany) by spin-down infection at 1400 rpm for 2 h; 1 μg/ml puromycin (Sigma-Aldrich, Germany) was used to select stable cell lines 3 days later. Three biologically independent replicates were carried out. Reverse-transcription quantitative real-time PCR (RT-qPCR) was performed to measure the knockdown effect of shRNA.
Total RNA was extracted from infected cells using the RNeasy Mini Kit (Qiagen, USA), and 1 μg of RNA was reverse transcribed using the PrimeScript RT reagent Kit with gDNA Eraser (Takara, Japan) and qPCR amplifications using TB Green Premix Ex Taq II (Takara, Japan). GAPDH was used as a reference gene (Table S3). Cell proliferation was detected using a Cell Counting Kit-8 (CCK-8) (Dojindo, Japan) in 96 well plates according to the manufacturer’s instructions. The cell cycle (CycleTest PLUS DNA Reagent Kit, catalog no. 340242) and the cell apoptosis (Annexin V-PE Apoptosis Detection Kit, catalog no. 559763) analysis were measured by BD flow cytometry according to the manufacturer’s instructions.
Statistical analysis
We used SPSS 24.0 (SPSS, Chicago) statistical software for statistical analysis. Comparisons of categorical variables were determined by Pearson’s chi-squared test or Fisher’s exact test. Two-sided P < 0.05 was considered statistically significant.
Results
Somatic mutations in newly diagnosed ALL patients
To better understand the landscape of somatic mutations in Chinese children with ALL, we performed targeted sequencing of 140 pediatric ALL patients (114 B-ALL and 26 T-ALL) with matched germline and diagnostic samples. The average sequencing coverage reached 634.06X (range 109.17X~ 1149.39X) and 128.43X (45.58X~ 500.37X), respectively, in tumor and control samples, which allowed accurate determination of mutant allele fractions and somatic mutation analysis (Figure S1A). In total, we detected 2193 somatic SNVs, 87 deletions and 56 insertions in the 950 sequenced genes. The average number of somatic mutations detected was 8.8 (range 0~78) per patient, including the nonsynonymous and synonymous mutations. We found no correlation between the number of mutations and gender, age, and initial white blood cell (WBC) counts. There was a trend towards more somatic mutations in T-ALL (average 8.0) than in B-ALL (average 6.0), although no significant difference (P = 0.267) was achieved plausibly because of limited sample size (Figure S1B). Basic characteristics of patients are described in Table S4.
Mutational spectrum analysis revealed that C > T single-base substitution was the dominant mutational event, which has been observed in all common cancer types and is likely caused by a spontaneous endogenous deamination process [22, 23]. By comparison, we found that B-ALL and T-ALL showed the highest rates of C > T (39.5%) and T > C (26%) substitutions, respectively (Figure S1C). The allele fractions (AFs) of SNVs were binomially distributed with a major peak around AF 0.15 (Figure S1D), suggesting a large fraction of somatic mutations were from the subclones.
Mutational landscape of pediatric ALL
To identify the somatic mutations of potential pathogenicity in ALL, we excluded several genes whose protein sequences and structural changes were not predicted to be deleterious (materials and methods). Based on 261 (25.9%) of the non-silent mutations predicted to be deleterious, we estimated the mutation prevalence and found that recurrently mutated genes with a mutation prevalence over 5% included KRAS (8.76%), NRAS (6.4%), FLT3 (5.7%) and KMT2D (5.0%) in childhood ALL. The most frequently mutated genes were members of the Ras signaling pathway (KRAS, NRAS, FLT3, NF1, PTPN11), especially in HeH, where 50% mutations occurred in the Ras pathway. In 30 HeH patients, FLT3 had the most recurrent mutations with a mutation prevalence of 20, and 75% FLT3 mutations occurred in HeH.
We observed obvious differences in terms of mutational landscape between B-ALL and T-ALL patients. The most frequently mutated genes were KRAS (11.4%), NRAS (7.0%), FLT3 (7.0%), and KMT2D (5.3%) in B-ALL, whereas NOTCH1 (23.1%), FBXW7 (23.1%), PHF6 (11.5%) and PTEN (11.5%) were enriched in T-ALL. The most prevalent mutations were enriched in the Ras signaling pathway (KRAS, NRAS, FLT3, NF1) and Notch pathway (NOTCH1, FBXW7) in B-ALL and T-ALL, respectively (Fig. 1). We also found that somatic mutations in the Ras signaling pathway displayed a similar pattern in which few mutations coexisted in patients with recurrent translocations (i.e., ETV6-RUNX1, BCR-ABL1, MLLr and TCF3-PBX1). However, mutations in the Notch signaling pathway were often shown in patients with fusions of SIL-TAL1 and MLLr (Table 1). When further calculating the number of pathogenetic genes within ALL subtypes, we found that two patients with intrachromosomal amplification of chromosome 21 (iAMP21) had a higher mutation burden (11.5/patient), however, the sample size is limited and the results need to be verified.

Mutational landscape of newly diagnosed 140 pediatric ALL patients. Heatmap diagram showing genomic data of 140 ALL patients, each of which is represented by a column, and each row represents a gene. Each color box indicates a type of mutation
Recurrently targeted pathways in pediatric ALL
Mutations in the Ras signaling pathway were more abundant in B-ALL
The most frequently mutated genes were members of the Ras signaling pathway, and Ras mutations were more abundant in B-ALL. The well-known hotspot mutations in the Ras genes included G12C/D/S/V (KRAS = 4; NRAS = 5), G13D/S/V (KRAS = 2; NRAS = 3), Q61K/H (KRAS = 1; NRAS = 1) and other mutational sites, such as A146T/P (KRAS = 3) and K117N (KRAS = 1). Interestingly, we found that one patient harbored both KRAS (G12C) and NRAS (G12D) mutations simultaneously with AFs less than 0.2, implying that at least two leukemia clones existed (Figs. 1 and 2a), however the patient with primary bone marrow blasts more than 97%.

Recurrent somatic mutations in diagnostic ALL patients
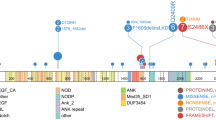
Schematic of protein structures showing mutations recurrently identified in diagnostic ALL samples. Proteins involved in the Ras pathway (a), Notch pathway (b), Epigenetic regulators (c) and cell cycle (d).
FLT3 plays a key role in hematopoietic cell growth and survival, which codes for a cell surface tyrosine kinase receptor. It was the most frequently altered gene in HeH in our research, and somatic mutations in FLT3 predominantly occurred in the tyrosine kinase domain and juxtamembrane domain, with the D835 residue as the most frequently mutated site [24]. Here, we identified several novel recurrent mutational sites in the kinase domain (D835A, Y842S, R845G) and in the transmembrane region (V592A, V592D, V592F), which may be involved in the regulation of FLT3 dimerization and self-activation. No FLT3-ITD mutations were detected in the entire cohort (Fig. 2a).
In addition, loss-of-function mutations in the Ras signaling negative regulator (NF1) occurred in 2 patients: R1306X (nonsense mutation) and R652 Vfs*36 (frameshift mutation). PTPN11 encodes a phosphatase that modulates signaling from upstream receptor tyrosine kinase and the Ras genes. In our cohort, we identified only a mutation (G60S) in PTPN11 reported as pathogenic in Noonan syndrome [25] in one patient. Janus kinase family members were also mutated, and novel JAK1 mutations were found in 3 patients (1 B-ALL and 2 T-ALL), S703I, D604Y and L910P.
Mutations in the notch signaling pathway were more common in T-ALL
In our cohort, T-ALL comprised 18.6% (n = 26) of ALL patients, and the most commonly mutated genes included NOTCH1 (23.1%), FBXW7 (23.1%), PHF6 (11.5%), PTEN (11.5%) and JAK1 (7.7%). The Notch signaling pathway, with the most common abnormality in T-ALL, has important roles in hematopoiesis, angiogenesis, cell proliferation, apoptosis and T cell development. We identified 6 NOTCH1 mutations, including 4 novel missenses (l1678P, A375G, R1598P and I1616N) and 2 frameshift mutations (Q1455 L fs*25, V2433G fs*35), with the majority of mutations in the heterodimerization domain (HD) (e.g., R1598P, I1616N and L1678P), which led to constitutive activation of the Notch pathway (Fig. 2b).
Six unique mutations in FBXW7, a component of the E3 ubiquitin ligase complex that controls protein turnover, occurred in 23.1% of T-ALL cases. The well-appreciated activating hotspot mutations R505C (two cases), D399G (one case) in the WD domain, and several novel mutations were identified (Fig. 2b). Notably, two cases (7.7%) included both NOTCH1 and FBXW7 mutations, and two cases included both NOTCH1 and PHF6 mutations. In addition, a hot spot of the in-frame deletion mutation at codon 6 in NOTCH2, another member of the Notch family, was observed in 4 cases (3 B-ALL, 1 T-ALL) (Fig. 2b).
Alterations in epigenetic regulations
Members of the histone methyltransferase MLL family were mutated frequently. KMT2A, known as myeloid/lymphoid or mixed-lineage leukemia (MLL), is a well-recognized leukemia-related gene and is rearranged in approximately 75% of infants with B-ALL, particularly in those less than 6 months of age [26]. However, the role of other MLL family members in hematological malignancy has not been fully established. In our cohort, we found that KMT2D, was the most frequently mutated epigenetic factor. Strikingly, KMT2D displayed a higher proportion of inactivating mutations (2 nonsense mutations, 4 frameshift mutations, and 2 missense mutations) (Fig. 3a). This result implied that inactivating mutations lead to a loss of function in a potential tumor suppressor.

KMT2D is a key oncogene in pediatric ALL. a Mutational diagram of KMT2D. PHD, plant homeodomain; HMG, high mobility group domain; SET, Su (var)3–9 Enhancer of zeste and Trithorax domain; FYR, FY-rich domain. b Increased KMT2D mRNA expression in ALL samples. **, P < 0.01. c Higher expression levels of KMT2D in ALL data (retrieved from GSE13159). d Generation of Nalm-6 cells with stable knockdown of KMT2D. Three shRNA sequences displayed significant suppression of KMT2D expression. ***, P < 0.001. e, f, g Evaluation of the effect of KMT2D knockdown Nalm-6 cells on cell proliferation, cell apoptosis and cell cycle. *, P < 0.05; **, P < 0.01; ***, P < 0.001, N.S., no significance. h Volcano plot depicting differentially expressed genes between the KMT2D knockdown and control groups. i Bubble chart depicts Gene Ontology (GO) functional enrichment analysis of differentially expressed genes. g RT-qPCR analysis of selected genes identified as differentially expressed in RNA sequencing. GAPDH was used as an endogenous control to normalize for RNA quality. *, P < 0.05; **, P < 0.01; ***, P < 0.001, N.S., no significance
However, the function of KMT2D in leukemia pathogenesis remains uncharacterized. By examining the gene expression in our patients and related ONCOMINE data (retrieved from GSE13159, the European Leukemia Network), we found that the KMT2D gene was highly expressed in both datasets (Fig. 3b, c) [27, 28]. To investigate the functional consequences of the loss-of-function mutations of KMT2D in ALL, we stably downregulated the expression of KMT2D in Nalm-6 cells using shRNA-mediated gene knockdown approach. We found that all 3 shRNA sequences significantly reduced the expression of KMT2D at the mRNA transcript levels (Fig. 3d) and that KMT2D knockdown cells exhibited a significant decrease in the cell numbers from day 4 (Fig. 3e). Consistently, downregulation of KMT2D promoted the apoptosis of Nalm-6 cells (early stage and late stage, Fig. 3f) and inhibited cell proliferation (significantly increased cell numbers in G0/G1 phase fraction and concomitant decreased in S phase fraction, Fig. 3g). Next, we performed RNA sequencing in both KMT2D knockdown and control Nalm-6 cells to examine transcriptomic changes caused by suppression of KMT2D. Significantly, 94 genes were upregulated in the KMT2D knockdown cells compared with the control cells, whereas 193 genes were downregulated (Fig. 3h). Gene ontology analysis revealed that differentially expressed genes were enriched in immune response, cell plasma membrane and T cell differentiation (Fig. 3i). Using quantitative real-time PCR, we validated the expression changes of several key genes involved in hematopoietic development and immune regulation, including POU2F2, TMPRSS3, TSPAN8, IL21R downregulated, and BCL6, ETV5, ZNF521, HSH2D upregulated in KMT2D knockdown cells (Fig. 3g, Table S3). Together, these findings underscored the critical role of the KMT2D gene in lymphoid malignancy and provided a potential therapeutic target for this cancer.
SETD2 mutations occurred in 2.6% of B-ALL cases and approximately 70% of the SETD2 lesions were likely to be loss-of-function mutations, including the nonsense mutation (C2525X), frameshift mutations (S165Lfs*12, A158Dfs*13, S1572Xfs*1) and splice site mutations (c.4715(exon5), c.4715 + 1(IVS5) ins TTTTATGAT) (Fig. 2c). Mutations in CREBBP occurred in 2 B-ALL patients at 2 new mutational sites (Y1450D, A1473T) (Fig. 2c) in the HAT domain. Inactivating mutations of PHF6 occurred in 3 T-ALL patients, and 2 cases with PHF6 mutations co-occurred with NOTCH1 mutations. However, EZH2 mutations in the catalytically active SET domain in 2 B-ALL patients, coexisting with ETV6 mutations (Figure S2). SUZ12 mutations occurred in only one T-ALL patient, with two types of somatic mutations, and coexisted with KMT2D and TP53 mutations, suggesting a potential interplay of these genes in the pathogenesis.
Transcription factors and cell-cycle pathway
Transcription factors ETV6 and PAX5 are essential for hematopoietic and lymphoid differentiation. In our cohort, ETV6 mutations were identified in 4 ALL cases, and PAX5 mutations were uncovered exclusively in 2 B-ALL cases (Table S5). Other mutated genes are mainly involved in the cell-cycle pathway, including TP53 and PTEN. TP53 mutations had 4 different types in its DNA-binding domain, including well-known hotspot R273H and other new mutations (Y205D, H179Mfs*68, S166X), these mutations occurred in TP53 DNA-binding domain may also inactivate TP53 by affecting its DNA-binding ability [29]. We also found 6 different mutations in PTEN (the tumor suppressor phosphatase and tensin homolog) in 3 T-ALL cases (Fig. 2d), as described tumor-associated mutations may occur in all PTEN domains.
In addition, other mutations, including NBPF10 (n = 7), MDC1 (n = 2) and CCND3 (n = 1), were also found in our research (Table S5). The majority of mutations were missense mutations and could be found in other studies, suggesting that these mutations also had significant meaning in ALL.
Discussion
In this study, we performed a genetic mutational analysis of Chinese children with ALL and identified an abundance of somatic mutations in essential genes, many of which were likely deleterious and may contribute to the pathogenesis of ALL. Although many of the most frequent mutations in pediatric ALL have been described previously, we identified distinct mutational characteristics and influenced different signaling pathways between B-ALL (Ras pathway) and T-ALL (Notch pathway) in this Chinese cohort. Ras pathway mutations were recurrent in pediatric B-ALL [24, 30, 31], and the vast majority of mutations occurred in KRAS, NRAS, FLT3 and NF1, revealing a central role of these genes in pediatric B-ALL. Ras genes mutational sites included G12C/D/S/V, G13D/S/V, Q61K/H, A146T/P and K117N, which were also identified in the study by Ding LW, et al. [32], suggesting that these mutational sites were common in Asian. one patient occurred KRAS and NRAS mutation simultaneously, these two mutations were close enough to be spanned by the same read-pair allowing the determination if the mutations are on either the same or different alleles [32]. We also found that 75% high-hyperdiploid possessed FLT3 mutations, which higher than 25% incidence as previous studies [33, 34], indicating a higher incidence in the Chinese patients with ALL associated with hyperdiploidy. Consistent with previous reports [20, 35], we also observed a high incidence of Ras pathway mutations in high-hyperdiploid patients with low mutation rates in TCF3-PBX1 and MLL rearrangement cases. Similar research was showed that B-ALL patients carrying any of the recurrent translocations ETV6-RUNX1, BCR-ABL or TCF3-PBX1 harbored few mutations compared to the other B-ALL patients [36]. Overall, this further underscores the crucial role of RAS mutations in ALL and highlight the genetic heterogeneity of pediatric ALL.
In our cohort, NOTCH1 mutations occurred in 23.1% of T-ALL cases, which was significantly lower than previously reported values [26, 29]. However, it is interesting that 2 cases with PHF6 mutations co-occurred with NOTCH1 mutations and were significantly correlated with the NOTCH1 mutation in Chinese adult T-ALL (PHF6 mutNOTCH1mut vs PHF6 wtNOTCH1mut, 75.0% vs 44.2%; P = 0.035) [37]. This discrepancy could be caused by the limited number of T-ALL cases enrolled in this study (n = 26), or possible coverage bias impairing ability to call gene sequence [38], and the detection of sequence mutations in ALL was insufficient. Frequently, some genes are affected by more than one type of alterations such as point mutation, copy number alterations (CNAs), focal aberrations/small insertions/deletions (INDEL), or structural variations (SVs). So, only one type of analysis lead to the underestimation of the mutation frequency of NOTCH1. Similarly, we underestimated the mutation frequencies of CDKN2A/2B, ETV6 and PAX5, due to lack of analysis of somatic copy number gains or losses. Copy losses of CDKN2A/2B (9p21), PAX5 (9p13) and ETV6 (12p13) were prevalent in children, while copy gains of RUNX1 (21q22.3) were more enriched in children [39]. So, large deletion, amplification and structural variant should be warranted; no single type of sequencing is capable of detecting the same alterations; WES is useful for point mutation investigation, whereas WGS can reveal SVs. Besides, NGS is increasingly being used to monitor drug response and treatment toxicity [40], contributing to the refinement of diagnosis and prognosis for 34% of patients with hematologic malignancies and blood disorders [41]. Incorporating pharmacogenomics and pharmacotranscriptomics can provide an enormous of molecular markers responsible for the efficacy, side effects, and toxicity of the chemotherapeutic drugs to improve the treatment protocols [42]. Then, utilizing genomic technology can better management and potential improve the survival rate in pediatric ALL patients.
In our findings, the most frequently mutated gene of epigenetic regulators was KMT2D, which encodes histone methyltransferase for methylates the Lys-4 position of histone H3, and its mutation can cause Kabuki syndrome, an autosomal dominant disease [43]. KMT2D is a key regulator of transcriptional enhancer function and plays an important role in maintaining genomic stability [44], and it is mutated in a large number of different cancers (e.g., diffuse large B cell lymphoma, small cell undifferentiated lung cancer, and medulloblastoma) [45,46,47]. As KMT2D is a predicted tumor driver gene in ALL [19] and it overexpressed in ALL, when KMT2D is knocked down, it significantly decreased leukemia cell growth, promoted cell apoptosis, and inhibited cell proliferation. A related study also showed that KMT2D was overexpressed in primary gastrointestinal diffuse large B cell lymphoma (PGI-DLBCL) and appeared as a prognostic factor for patients older than 60 years old [48]. KMT2D overexpression was observed in esophageal squamous cell carcinoma (ESCC), predicting poor clinical outcomes and facilitating ESCC tumor progression [49]. In addition, KMT2D can interact with KMT2A in acute myeloid leukemia, its deletion reduced MLL-AF9 leukemia cell survival, and the codeletion of both KMT2A and KMT2D resulted in more severe reductions in survival, proliferation, and gene expression than either individual gene deletion [50]. Hence, the KMT2D gene plays an important role in hematological tumors and may act as a drug target in MLL-rearranged leukemia. However, there existed limitation in our research, the off-target effect remains one of the major obstacles in KMT2D-shRNA experiment and it is insufficient to research the function of KMT2D in ALL. So, we should generate a KMT2D knock-out cells by CRISPR-Cas9-mediated genome editing to demonstrate its potential molecular pathogenesis in ALL in the future study.
As the main part of this study, we intend to show the genomic landscape of pediatric ALL from a single center in China, and our results provided a substantial number of genetic variants contributing to accumulate genetic data of Chinese children and explore molecular determinants in the future. However, there are some limitations in the present study. The number of patients enrolled in the present study was limited, and sample selection may be biased, which may contribute to the discrepancies in the findings between our study and others, and collaborative efforts with larger sample sizes are needed. Structural alterations may play important roles in leukemogenesis; thus, the absence of this information leads to incomplete understanding of the genetic basis of ALL. More comprehensive approaches, such as WGS, RNA-seq, pharmacogenomics and pharmacotranscriptomics, and larger integrative studies, can be warranted to dissect the underlying complexity of ALL in the future. The frequencies and distributions of abnormalities of ALL patients between children and adult, Chinese and western should further be compared in a larger cohort.
Conclusion
This study provided further insights into the genetic basis of ALL and strengthened that Ras mutations were predominant in childhood ALL, especially in the subtype of high-hyperdiploid. These findings have major implications for understanding the genomic complexity of ALL and also have direct implications for the clinical management of ALL.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request for privacy reasons.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- B-ALL:
-
B cell ALL
- FISH:
-
Fluorescence in situ hybridization
- HeH:
-
High hyperdiploid
- iAMP21:
-
Intrachromosomal amplification of chromosome 21
- MAF:
-
Minor allele frequency
- MLL:
-
Mixed-lineage leukemia
- NGS:
-
Next-generation sequencing
- RNA-seq:
-
RNA sequencing
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- shRNAs:
-
Small hairpin RNAs
- SNPs:
-
Single-nucleotide polymorphisms
- SNV:
-
Single-nucleotide variant
- T-ALL:
-
T cell ALL
- WBC:
-
White blood cell
- WES:
-
Whole exon sequencing
- WGS:
-
Whole genome sequencing
References
Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541–52.
Wiemels JL, Cazzantiga G, Daniotti M, Eden OB, Addison GM, Masera G, et al. Prenatal origin of acute lymphoblastic leukemia in children. Lancet. 1999;354:1499–503.
Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukemia. Nat Rev Cancer. 2003;3:639–49.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–52.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature. 2007;446:758–64.
Inaba H, Geaves M, Mullighan CG. Acute lymphoblastic leukemia. Lancet. 2013;381:1943–55.
Chiaretti S, Gianfelici V, Ceglie G, Foà R. Genomic characterization of acute leukemia. Med Princ Prac. 2014;23:487–506.
Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12:344–57.
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45:1226–31.
Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47:180–5.
Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukemia. Nat Commun. 2015;6:6604.
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukemia. Nature. 2011;471:235–9.
Malinowska-Ozdowy K, Frech C, Schönegger A, Eckert C, Cazzaniga G, Stanulla M, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29:1656–67.
Tzoneva G, Perezgarcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–71.
Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–4.
Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21:563–71.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating Leaions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15.
Chalandon Y, Thomas X, Havette S, Cayuela JM, Abbal C, Huguet F, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. 2015;125:3711–9.
Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, et al. The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum Mutat. 2015;36:118–28.
Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47:672–6.
Chen B, Wang Y-Y, Shen Y, Zhang WN, He HY, Zhu YM, et al. Newly diagnosed acute lymphoblastic leukemia in China (I): abnormal genetic patterns in 1346 childhood and adult cases and their comparison with the reports from Western countries. Leukemia. 2012;26:1608–16.
Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84.
Alexandrovi LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–7.
Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Nati Acad Sci US. 2016;113:11306–11.
Ezquieta B, Santome JL, Carcavilla A, Guillen-Navarro E, Perez-Aytes A. Sanchez del Pozo J, et al. alterations in RAS-MAPA genes in 200 Spanish patients with Noonan and other neuro-cardio-facio-cutaneous syndromes. Genotype and cardiopathy. Rev Esp Cardiol. 2012;65:447–55.
Tasian SK, Hunger SP. Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. Br J Haematol. 2017;176:867–82.
Kohlmann A, Kipps TJ, Rassenti LZ, Downing JR, Shurtleff SA, Mills KI, et al. An international standardization program towards the application of gene expression profiling in routine leukemia diagnostics: the microarray innovations in LEukemia study prophase. Br J Haematol. 2008;142:802–7.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the international microarray innovations in leukemia study group. J Clin Oncol. 2010;28:2529–37.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8.
Ariës IM, Van den Dungen RE, Koudijs MJ, Cuppen E, Voest E, Molenaar JJ, et al. Towards personalized therapy in pediatric acute lymphoblastic leukemia: RAS mutations and prednisolone resistance. Haematologica. 2015;100:e132–6.
Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–30.
Ding LW, Sun QY, Tan KT, Chien W, Mayakonda A, Yeoh AEJ, et al. Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. 2017;77:390–400.
Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103:3544–6.
Braoudaki M, Karpusas M, Katsibardi K, Papathanassiou C, Karamolegou K, Tzortzatou-Stathopoulou F. Frequency of FLT3 mutations in childhood acute lymphoblastic leukemia. Med Oncol. 2009;26:460–2.
Jerchel IS, Hoogkamer AQ, Ariës IM, Steeghs EMP, Boer JM, Besselink NJM, et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia. 2018;32:931–40.
Lindqvist CM, Lundmark A, Nordlund J, Freyhult E, Ekman D, Almlof JC, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. 2016;7:64071–88.
Li M, Xiao L, Xu J, Zhang R, Guo J, Olson J, et al. Co-existence of PHF6 and NOTCH1 mutations in adult T-cell acute lymphoblastic leukemia. Oncol Lett. 2016;12:16–22.
He J, Abdelwahab O, Nahas MK, Wang K, Rampal RK, Intlekofer AM, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14.
Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–83.
Heikamp EB, Pui CH. Next-generation evaluation and treatment of pediatric acute lymphoblastic leukemia. J Pediatr. 2018;203:14–24 e2.
Marks LJ, Oberg JA, Pendrick D, Sireci AC, Glasser C, Coval C, et al. Precision medicine in children and young adults with hematologic malignancies and blood disorders: the Columbia University experience. Front Pediatr. 2017;5:265.
Pavlovic S, Kotur N, Stankovic B, Zukic B, Gasic V, Dokmanovic L. Pharmacogenomic and Pharmacotranscriptomic profiling of childhood acute lymphoblastic leukemia: paving the way to personalized treatment. Genes (Basel). 2019;10:191.
Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies MLL2 mutations as a cause of kabuki syndrome. Nat Genet. 2010;42:790–3.
Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016;30:408–20.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95.
Ross JS, Wang K, Elkadi OR, Tarasen A, Foulke L, Sheehan CE, et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol. 2014;67:772–6.
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9.
Ye H, Lu L, Ge B, Gao S, Ma Y, Liang B, et al. MLL2 protein is a prognostic marker for gastrointestinal diffuse large B-cell lymphoma. Int J Clin Exp Pathol. 2015;8:13043–50.
Abudureheman A, Ainiwaer J, Hou Z, Niyaz M, Turghun A, Hasim A, et al. High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2018;144:1025–35.
Chen Y, Anastassiadis K, Kranz A, Stewar AF, Arndt K, Waskow C, et al. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell. 2017;31:755–70.
Acknowledgements
We thank all patients and their families who participated in this study and we also would like to thank all of our colleague for their contribution to this study.
Funding
The research was funded by the Research Programs of the Shanghai Science and Technology Commission Foundation (No.14411950603), Shanghai Municipal Commission of Health and Family Planning (No. 201740011), and Project Ai You Foundation Supporting Children with Cancer Program. The funding bodies were not involved in the design of the study, in the collection, analysis, and interpretation of the data, or in writing of the manuscript.
Author information
Authors and Affiliations
Contributions
XZ is the principal investigator and takes primary responsibility for the paper. HW contributed to the conception and design of the study. XQ acquired and managed the patient samples. YC and JM performed the DNA and RNA extraction. SG was involved in sample and library preparation for targeted sequencing. HZ performed experiments using cell models, and JL helped to conduct flow cytometry. HZ and JX performed the data integration and analysis. HZ drafted the paper; HW helped interpret the data and contributed to the critical revision of the manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The written consent was obtained from the patients’ parents or legal guardians in accordance with the Declaration of Helsinki and the study was approved by the Institutional Review Board of the Fudan Institutes, Shanghai, China.
Consent for publication
Not applicable.
Competing interests
The author declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
950 Genes screened in the exon sequencing. Table S2. Sequences of shRNA used in this study. Table S3. Sequences of real-time PCR primers used in this study. Table S4. Clinical characteristics and genetic types of patient cohorts. Table S5. Other mutations occurring in our ALL cohort. Figure S1. Somatic mutations in acute lymphoblastic leukemia (ALL). A, Boxplots showed the median depth of coverage depth in tumor samples and the control samples (matched germline samples). B, Boxplots showed the median number of somatic mutations detected in B-ALL and T-ALL. C, Pattern of single base substitution in B-ALL and T-ALL patients. D, Density plots of the allele fraction (AF) of single nucleotide variants (SNVs) in the B-ALL and T-ALL patients. The main clones with a maximum AF close to 0.4 and subclonal mutations with a maximum AF below 0.25. Figure S2. Recurrent mutations in epigenetic regulations. Schematic diagrams of protein structures involving gene mutations in PHF6, EZH2, SUZ12.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Wang, H., Qian, X. et al. Genetic mutational analysis of pediatric acute lymphoblastic leukemia from a single center in China using exon sequencing. BMC Cancer 20, 211 (2020). https://doi.org/10.1186/s12885-020-6709-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-6709-7