
Abstract
Background
ARID1A has been described as a tumor suppressor gene, participating in chromatin re-modeling, epithelial-mesenchymal-transition and many other cellular and molecular processes. It has been cited as a contribute in tumorigenesis. The role of ARID1A in CRC is not yet defined.
Aim
To investigate the role of ARID1A methylation and CNV in its expression in CRC cell lines and to examine the relationship between ARID1A status with survival and clinicopathologic characteristics in patients with CRC.
Methods
We used RT-PCR to determine both CNV and expression of ARID1A from six CRC cell lines. We used MSP to evaluate methylation of ARID1A. IHC was used to assess ARID1A protein expression. We also evaluated MSI and EMAST status in 18 paired CRC and adjacent normal tissues. 5AzadC was used to assess effect of DNA demethylation on ARID1A expression. Statistical analysis was performed to establish correlations between ARID1A expression and other parameters.
Results
Among the 18 CRC tumors studied, 7 (38.8%) and 5 tumors (27.7%) showed no or low ARID1A expression, respectively. We observed no significant difference in ARID1A expression for overall patient survival, and no difference between clinicopathological parameters including MSI and EMAST. However, lymphatic invasion was more pronounced in the low/no ARID1A expression group when compared to moderate and high expression group (33% VS. 16.6% respectively. ARID1A promoter methylation was observed in 4/6 (66%) cell lines and correlated with ARID1A mRNA expression level ranging from very low in SW48, to more pronounced in HCT116 and HT-29/219. Treatment with the methyltransferase inhibitor 5-Azacytidine (5-aza) resulted in a 25.4-fold and 6.1-fold increase in ARID1A mRNA expression in SW48 and SW742 cells, respectively, while there was no change in SW480 and LS180 cells. No ARID1A CNV was observed in the CRC cell lines.
Conclusion
ARID1A expression is downregulated in CRC tissues which correlates with it being a tumor suppressor protein. This finding confirms ARID1A loss of expression in CRC development. Our in-vitro results suggest high methylation status associates with reduced ARID1A expression and contributes to CRC tumorigenesis. However, there was no significant association between ARID1A loss of expression and clinicopathological characteristics. Future in-vivo analysis is warranted to further establish ARID1A role in colorectal neoplastic transformation.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is rapidly increasing in prevalence among several regions of the world, such as Asia [1, 2]. CRC is the second and the third major reason for cancer-related death among men and women, respectively [3, 4]. The molecular pathogenesis of CRC is driven by successive acquisition of epigenetic and genetic changes, which associate with silencing of tumor suppressor genes and activation of proto-oncogenes [5]. The low efficiency of conventional therapies to increase longevity in CRC patients, calls for new targeted treatment. Characterization of genes related to the progression and development of colon cancer showed that their clinical implication is crucial for diagnosis and efficient therapy.
ARID1A encodes the AT-rich interactive domain-containing protein 1A, a representative of the DNA-binding protein family and principal subunit of the SWI–SNF complex (switch/sucrose non-fermentable). ARID1A is frequently deleted in multiple human tumors. It is located on chromosome 1p36.11, a region that is commonly deleted in various cancer types and suspected to contain tumor suppressor genes [6,7,8,9,10]. For example, deletion of 1p36 region, harboring ARID1A, was observed in more than half pancreatic carcinomas [11, 12]. SWI–SNF is a highly conserved chromatin rearranging complex, applying adenosine triphosphate (ATP)-dependent helicase functions to facilitate the access of transcriptional repressors and activators to DNA [13, 14]. Therefore, the protein interferes with regulation processes, including development, differentiation and DNA repair [15]. ARID1A protein is typically deficient in oncogenic tissues, and therefore is suspected to possess the tumor suppressor activity of the complex [16, 17]. Also, ARID1A plays a critical role in the FAS-mediated apoptosis [18]. The interplay between gene and promoter is associated with the inhibition of cell-cycle-specific genes [19]. ARID1A knock-out cells do not enter cell cycle arrest [20]. The ARID1A involvement in cell-cycle arrest shows that it significantly aids tumor repression, through the SWI/SNF complexes [17]. A wide range of cancer-gene mutations are detected by NGS and loss of function mutations in the ARID1A is detected repeatedly and frequently in various cancer types [13, 21]. A recent study revealed that ARID1A knockdown in renal cells led to epithelial-mesenchymal-transition, highlighting its role in cell differentiation and tissue homeostasis [22].
Although ARID1A expression loss has been described chiefly in gynecological cancers, it is reported among other tumor types, such as from gastrointestinal tract tumors [23,24,25]. In gastric and gynecological cancers, ARID1A mutation or loss of ARID1A protein expression strongly correlates with microsatellite instability, and is inversely correlated with alterations in TP53 [23, 26]. Recently, there has been a growing interest in clinical significance of ARID1A low expression in gastrointestinal oncogenic conditions, particularly tumors manifesting DNA mismatch repair (MMR) deficiency [27].
Molecular mechanisms related to low ARID1A expression seem to be distinct amongst different cancerous tumors. For instance, ARID1A copy number loss is the major cause of low ARID1A expression in pancreatic cancer (47%) [28]. Prior studies indicate that copy number loss exists in 13–35% of breast cancers [17]. Mutation can also affect the expression in ovarian clear cell carcinoma, with ARID1A mutation (with 50% mutation rate) as the major cause of loss of expression. Moreover, among breast cancers, promoter hypermethylation and histone modification are the main reasons for loss of ARID1A expression [17].
Studies on ARID1A expression in CRC is limited. A relatively high mutation rate of ARID1A was reported in the colorectal cancer (10–40%) [13, 29,30,31], but it is not apparent whether DNA hypermethylation, and/or copy number variation (CNV) also are contributory in alteration of ARID1A expression. To explore the major molecular mechanisms of ARID1A expression loss in CRC, we aimed to study ARID1A methylation, expression and CNV in clinical samples and CRC cell lines. We also determine if treatment of these cell lines with 5-aza affect the expression of ARID1A. In addition, we examined possible correlations between ARID1A expression loss and various clinicopathological parameters in CRC tissues.
Materials and methods
Patient
Eighteen paraffin-embedded patient-derived paired CRC and adjacent nontumorous tissue samples were collected from the archives of the department of pathology of Faghihi Hospital of Shiraz University of Medical Sciences. All patients underwent primary tumors resection between 2016 and 2017. None of the patients had preoperative radiotherapy or preoperative chemotherapy. Tumor staging was determined according to AJCC TNM system. All tumors were histologically classified based on World Health Organization criteria. Clinical, pathologic and follow-up information of patients were obtained from hospital medical records. Overall survival (OS) was defined as the time interval (in months) from surgery to the time of death from any cause or to end of follow-up if the patient was alive (censored). Twelve separate cancer-matched normal pairs were from Howard University Hospital and used for exome sequencing [32]. The IRB committee of the Medical University of Shiraz and Howard University approved this study and the archival tissue were obtained, de-identified prior to receipt and there is no access to the identifiers (IRB-06-MED-39).
Immunohistochemistry and scoring
Immunohistochemistry was performed on 4-μm thick paraffin-embedded tissue sections from patients with colorectal cancer using a rabbit anti-human ARID1A antibody (HPA005456, Sigma, USA) at a dilution of 1:200. Briefly, sections were deparaffinized using xylene and rehydrated in a descending series of alcohol dilutions. Activity of endogenous peroxidase was inhibited with 3% hydrogen peroxide in methanol for 5 min. After, the sections were heated for 15 min at 120 °C in 10 mM citrate buffer for antigen retrieval. Sections were then blocked with goat serum 1:100 in PBS for 20 min and subsequently incubated with primary antibodies for 30 min at room temperature. After washing, immunohistochemical staining was performed using Master Polymer Plus Detection System kit (Incl. DAB Chromogen) according to the manufacturer’s instructions. Sections were then counterstained with hematoxylin, dehydrated by ethanol and mounted.
Immunostained sections were reviewed and scored by an expert pathologist who was blinded to patient’s clinicopathological and molecular information, according to the percentage of the stained cells and the intensity of staining. The percentage of positively stained cells was scored as: ≤ 10% = 0, 11–25% = 1, 26–50% = 2, 51–75% = 3, and > 75% = 4. Staining results were considered positive for tumor tissues with nuclear staining. Staining intensity was scored as negative = 0, weak = 1, moderate = 2, and strong = 3. The final score of each section equals the percentage score × intensity score. Based on the overall score, the immunostained sections were further divided into two groups: The group of ARID1A-negative (overall score = 0) and the group of ARID1A-positive (overall score ≥ 1). The group of ARID1A-positive was further divided into three subgroups: low ARID1A expression group (overall score = 1–4), moderate ARID1A expression group (overall score = 5–8) and high ARID1A expression group (overall score = 9–12).
MSI and EMAST analysis
MSI and EMAST analysis using DNA extracted from 18 paraffin-embedded patient-derived paired CRC and adjacent nontumorous tissue samples were performed as described previously [33, 34]. Briefly, MSI and EMAST status were analyzed using the standard panel for MSI detection in CRC samples (NR21, NR22, NR27, BAT25 and BAT26) and five EMAST markers (D9S242, D8S321, D20S82, D20S85 and MYCL1), respectively [35]. Microsatellite loci were amplified via PCR with previously described primers [33, 34]. Samples were classified as microsatellite instability high (MSI-H), microsatellite instability low (MSI-L), or microsatellite stable (MSS) based on the number of markers displaying instability and corresponding to ≥2 markers, one, or none, respectively. Samples were labeled EMAST-positive when at least two of the 5 the analyzed non-mononucleotide markers were unstable.
Cell cultures
In this study, six human CRC cell lines (HCT116, HT-29/219, SW48, SW742, SW480 and LS180) were purchased from the National Cell Bank of Iran (Pasteur Institute, Tehran, Iran). cells were cultured in RPMI 1640 (HCT116, SW48, SW742, HT29/219 and SW480) or DMEM (LS180), containing 10% fetal bovine serum, 1% penicillin, 2 mM glutamine, and 1% streptomycin, in a humidified CO2 (5%) incubator at 37 °C.
5-Aza-C treatment
Cells were cultured in T-25 flasks, after 24 h, cells were covered for 72 h with 2.5 μM 5-Azacytidine (5-aza) (Sigma), a methyltransferase inhibitor. The culture media were replaced every 24 h with fresh media, containing 5-aza. Stock solutions of 5-aza were prepared fresh every day, sterilized by filtration, and kept at 0 °C, until further use. For drug treatment, 5-aza was dissolved in acetic acid: water (1:1 v/v). Then, the stock solution (100 mM) was diluted, up to a final concentration of 3 μM in PBS and the cells treated with 5-aza for 3 day.
Extraction of DNA and methylation analysis
Genomic DNA was extracted from cells as follows: Cells were collected and initially lysed in 1.0 ml DNA lysis buffer (0.5% Triton X-100, 10 mM EDTA, 50 mM HEPES at pH 8), supplemented with 100 μl 1% SDS and 200 μg/mL proteinase K (Sigma-Aldrich). Following the addition of RNAse A (100 μg/mL), samples were incubated at 65 °C for 1 h. Subsequently, an equal volume of phenol:chloroform: isoamyl alcohol (25:24:1 v/v), was added to the cell lysate, shaken vigorously, and then spun at 12000 rpm for 10 min. Afterward, the supernatant was collected, and the isolated DNA was precipitated by adding 2 volumes of 100% ethanol and sodium acetate, at a concentration of 300 μM, and kept at − 20 °C for 24 h. The DNA was next dissolved in 50 μl of DNAse-free water and quantified with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA). The methylation status of CpG islands in the ARID1A promoter region was analyzed, using MS-PCR as previously described [36]. Briefly, the method detects methylation of bases in a bisulfite reaction, and subsequently detects them by polymerase chain reaction (PCR), using specific primers designed for bisulfite-converted methylated and unmethylated sequences.
The primer sequences used for the detection of ARID1A promoter region included: methylated, forward: 5′-GGCGTAGGTTTTAGAGATGC-3′, and reverse: 5′- AAACGAACTCGCTCCCTT-3′; unmethylated, forward: 5′-GGTGTAGGTTTTAGAGATGT-3′; and reverse: 5′ - AAACAAACTCACTCCCTT- 3′. The MS-PCR primers were designed using a methylation specific software (www.mspprimers.org). PCR reactions were performed in triplicates, using the following conditions: 95 °C for 10 min, and 35 cycles of 95 °C for 40 s, 59 °C (unmethylated DNA) or 53 °C (methylated DNA) for 40 s, and 72 °C for 30 s, followed by an extension at 72 °C for 10 min. A band size of 163 bp was observed in the ARID1A methylation set (Fig. 1). Distance of the 5′ nucleotide of the sense primers of methylated and unmethylated from the transcription start site (TSS) of the ARID1A was − 500 and − 498, respectively. The PCR amplicons were electrophoresed on 2% agarose gel, dyed with GelRed (Biotium, Belgium) and visualized, using the UV illumination system. Genomic DNA extracted from peripheral blood leukocytes, served as an unmethylated control sample. Human methylated DNA from Zymo Research (ZYMO Research, Freiburg, Germany), was used as methylated positive control. Distilled water was used as a blank control for the PCR reaction.

Schematic structure of the ARID1A CpG island, and the MSP region indicated. Each vertical line represents one CpG site. The transcription start site (TSS) is defined as position + 1, and the rest of the sequence is numbered relative to the TSS
RNA extraction and reverse transcription
Total RNA was extracted from sub-confluent cell cultures, using the Tripure RNA isolation kit (Roche Applied Science, Germany), based on the manufacturer’s guidelines. The isolated RNA was quantified by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). Furthermore, RNA integrity was examined by the presence of the 28S, 18S rRNA bands on 1.5% agarose gel. The cDNA was synthesized, with DNase-treated RNA, 2 μg of RNase-free, random primers or random hexamers, using the RevertAid First Strand cDNA Synthesis Kit (Fermentas), in 20 μl volumes reaction, based on the manufacturer’s instructions.
Analysis of gene expression
Real-time quantitative PCR (qPCR) was carried out, using ABI 7500 Sequence Detection System (Applied Biosystems, USA) and the following primers for the ARID1A mRNA analysis: forward, 5′-CAGTACCTGCCTCGCACATA-3′; reverse, 5′ GCCAGGAGACCAGACTTGAG- 3′. Additionally, GAPDH was used as the internal housekeeping gene with the following primers: forward, 5′- CGACCACTTTGTCAAGCTCA- 3′, reverse, 5′- AGGGGTCTACATGGCAACTG-3′. The PCR reaction mixture contained 1 μl cDNA of each sample, 1 μl of forward and reverse primers (0.2 μmol L-1 final concentration) and 12.5 μl SYBR Green master mix (ABI, UK) in a total final volume of 25 μl. PCR was carried out using the following conditions: a pre-cycling thermal activation at 95 °C for 15 min, and 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 30 s, extension at 72 °C for 30 s, and a final extension of 72 °C for 10 min, followed by a melt curve (58–95 °C) analysis. Experiments were performed in duplicate. Furthermore, products specificity was confirmed by electrophoresis on 2% agarose gel. The relative expression was calculated, using the 2-ΔΔCT method and normalized to the GAPDH expression.
Copy number variation analysis of ARID1A
Real time quantitative PCR (qPCR) was carried out to reveal the relative changes in ARID1A copy numbers among six CRC cell lines and a normal colon tissue sample. Ten nanograms of genomic DNA from each cell line and normal colon tissue was used as template to carry out the real time PCR reaction, using specific primer for ARID1A, and RNase P (two copies in a diploid genome) as a reference gene. Primers used for the RNase P are as follows: Forward 5′-AGATTTGGACCTGCGAGCG-3′ and reverse 5′- GAGCGGCTGTCTCCACAAGT-3′, giving an amplicon size of 65 bp. Primers used for ARID1A are forward, 5′- GGATGGCACTTTGAGAAC − 3′ and reverse 5′- GAAAGATGAACAGGAGGA − 3′, giving a fragment length of 166 bp. Amplification of the product was carried out with the following conditions: a pre-cycling thermal activation at 95 °C for 15 min, accompanied by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension of 72 °C for 10 min. Experiments were performed in triplicates.
For all experiments, cycle threshold (Ct) values are extracted, with 7500 software (Applied Biosystems). Relative CNVs were computed, using the 2-ΔΔCT method, which were normalized to a calibrator sample (normal sample) to have normal diploid genomes) and a reference gene (RNase P). In comparison to the normal sample, a two-fold decrease or increase in the copy number of ARID1A in the CRC cell lines was referred to as a deletion or amplification, respectively.
Statistical analysis
Data analysis was performed, using SPSS software (Version 23). Data are presented as mean ± SD. The association between ARID1A expression and the clinicopathological characteristics were evaluated by Pearson χ2 test and Fisher’s exact test based on the type of data. The Kaplan-Meier method was used for survival rates estimation. Other experiments were analyzed, using the one-way ANOVA, accompanied by Tukey’s multiple comparison tests. Differences with a P value ≤0.05 were statistically significant.
Results
Differential expression of ARID1A in CRC cell lines
The ARID1A mRNA expression was examined in six colorectal cancer cell lines, SW48, SW742, HT29/219, HCT116, LS180, and SW480 (Fig. 2). ARID1A mRNA average levels (normalized to GAPDH mRNA), showed that SW48 cells expressed at the lowest levels. As shown in Figs. 2 and 3c, ARID1A mRNA expression was not observed in SW48 cells and therefore were used as baseline/reference for ARID1A expression levels (set at 1.0). The average expression level of the ARID1A mRNA in HCT116 cells was significantly higher (364 fold; P < 0.05) in comparison with the other cell lines. Quantitation of the ARID1A mRNA revealed that HT29/219, LS180, SW480 and SW742 cells, expressed approximately, 136.8, 24.3, 15.5 and 8.6- fold more than the SW48 cells, respectively. We observed a spectrum of high-level expression (in HCT116) to virtually no expression (in SW48) for ARID1A mRNA (Fig. 2).

ARID1A relative mRNA expression in SW742, LS180, HCT116, HT29/219, SW480 and SW48 cell lines was determined by Real-Time RT-PCR. Each gene’s expression values was normalized to the GAPDH mRNA. SW48 cell line was used as a reference, with an expression level set to 1.0, and expressions in all other cell lines were presented as an n-fold difference, compared to the SW48. Mean ± SD of three experiments is reported (p < 0.05)

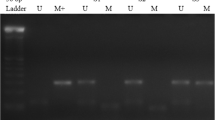
Methylation and mRNA expression status of ARID1A before and after treatment with 5-aza in CRC cell lines. a Methylation status of the ARID1A promoter was examined by MSP in CRC cell lines. The methyltransferase inhibitor (5-Aza) induced demethylation in CRC cell lines by MSP, U: unmethylated, M: methylated. b Treatment with 5-Aza alters the ARID1A mRNA expression in a cell line-dependent manner. Relative ARID1A mRNA expression before and after 5-Aza treatment were examined by Real-Time RT-PCR. ARID1A mRNA expression was normalized to GAPDH mRNA. Demethylation treatment restored ARID1A mRNA expression in SW48, SW742, LS180, HT29 cell line by RT-PCR. The data were presented as average fold-changes ± SD. cARID1A mRNA expression is silenced in SW48 cell line
ARID1A MSP analysis shows different methylation level in CRC cell lines
Since aberrant methylation plays a key role in regulating ARID1A expression in several cancers [17], we examined the methylation status of the ARID1A promoter in the six CRC cell lines by methylation specific PCR (MSP). MSP showed that SW48 cells were fully methylated (Fig. 3a); completely downregulating ARID1A mRNA expression. No ARID1A promoter methylation was detected in the HCT116 and HT29 cell lines (Fig. 3a). SW742, SW480 and LS180 cell lines displayed both methylated and unmethylated alleles (Fig. 3a). Coincident with HCT116 cells expressing the highest level of ARID1A (Fig. 2), there was no promoter methylation; SW48 cell matched ARID1A transcript downregulation with full methylation of the ARID1A promoter.
Expression analysis of ARID1A after treatment with 5-Aza-C
To determine if aberrant promoter methylation is the process affecting ARID1A mRNA expression, cells were treated with the DNA methyltransferase inhibitor 5-aza, and ARID1A expression was examined by real-time RT-PCR. SW48 cells that has full methylation dramatically restored ARID1A expression (25.4 fold) after 5-aza treatment (Fig. 3b and c) while HCT116 cells that had no ARID1A promoter methylation, did not show ARID1A expression changes (Fig. 3b). SW742 cells that show partial methylation, increased ARID1A expression by 6.1-fold with 5-aza treatment (Fig. 3a, b). Treating SW48, SW742, LS180 and SW480 cells with 5-aza resulted in an increase in the unmethylated PCR product due to reduction of ARID1A methylation but no overall mRNA expression (Fig. 3a and b).
Colorectal cancer cell lines show unaltered ARID1A copy numbers
During carcinogenesis, the gain or loss of ARID1A copy numbers could be the basis for variations in gene expression. In order to examine whether differences in basal ARID1A expression, was the consequence of ARID1A copy number variations, we employed real time PCR. Colorectal cancer cell lines, SW48, SW742, HT29/219, HCT116, LS180 and SW480 cells were analyzed with a normal DNA sample of colon tissue. The ARID1A copy numbers were normalized and calculated relative to the endogenous reference gene RNase P, which is shown to be available in two copies in a diploid genome.
The comparison between ARID1A copy number of CRC cell lines related to control revealed that CRC cell lines had no significant difference in ARID1A copy number (Fig. 4). These data suggest that the CNV is unlikely to be involved in the differences in ARID1A expression in the studied CRC cell lines.

Analysis of ARID1A copy number variation in the CRC cell lines. CRC cell lines: LS180, SW742, HCT116, HT29/219, SW480 and SW48 were analyzed relative to a DNA sample of a normal colon tissue, regarding the copy number variation (CNV) of ARID1A, using real-time PCR. In the CRC cell lines, ARID1A copy numbers were comparable with the control
ARID1A expression was frequently reduced in human colorectal cancer tissues
Immunohistochemistry was conducted on 18 paired normal CRC samples. Expression of ARID1A in adjacent nontumor mucosal epithelial cells was present in all patients. IHC analysis revealed that ARID1A protein expression was negative in 7 out of 18 (38.8%) tumor tissues when compared with adjacent non-malignant counterparts. Five (27.7%) tumors had low expression of ARID1A, 4 (22.2%) tumors had moderate expression of ARID1A, and 2 (11.1%) tumors had high expression of ARID1A. Thus, a total of 12 of 18 (66.6%) CRCs demonstrated negative or low ARID1A expression when compared with normal tissues. Collectively, these results demonstrate that ARID1A is frequently down regulated in CRC tumors. Typical immunostaining of high, moderate, low and negative expression of ARID1A are shown in Fig. 5.

Representative images of H&E and IHC of ARID1A from four CRC tumors. a Negative expression; b Low expression; c Moderate expression; and d High expression of ARID1A
ARID1A expression is not associated with clinicopathological parameters and survival rates in patients
Relationships between ARID1A expression and clinicopathological features was analyzed in patients with colorectal cancer. Table 1 summarizes the clinicopathologic data of the 18 CRC patients with statistical results. Among the 18 CRC patients tested, six (33.33%) were MSI-L/EMAST, five (27.7%) were MSI-high and the other seven (38.8%) were MSS. Loss of ARID1A expression was more common in CRCs arising in rectum (71.42% of all ARID1A-negative tumors vs. 54.54% of all ARID1A-positive tumors) and female (42.85% of all patients with loss of ARID1A vs. 27.28% of all patients without loss of ARID1A) and in association with both negative lymph vascular invasion and TNM stage III (71.42 and 57.14% of all ARID1A-negative tumors, respectively). Overall, MSI and MSI/EMAST are less represented in the no and low ARID1A expression group while lymphatic penetration seems to be more prominent in comparison with moderate and high expression group (33% VS. 16.66% respectively; p = 0.457). These trends failed to reach statistical significance. Overall, there was no significant association between the loss of ARID1A expression and any clinicopathological parameters examined.
In addition, we further evaluated the relationship between ARID1A expression and overall survival in colorectal cancer patients. The mean duration of follow up was 29.18 months, ranging 12–44 months, during which 2 patients died of the disease. Kaplan-Meier survival analysis showed that overall survival was worse for patients with positive ARID1A expression than the ones with negative ARID1A expression, although it was not statistically significant (Fig. 6). The Kaplan-Meier curve highlights the need for a larger number of patients to confirm the prognostic value of ARID1A expression in colorectal cancer.

Kaplan-Meier curves. Kaplan-Meier curves with log-rank test for patients with positive ARID1A expression vs. negative ARID1A expression
Exome data analysis from 12 pairs of cancer-matched normal tissues revealed that only one patient had mutations in ARID1A. The patient was KRAS wild type and displayed mutations in MSH6 and PKHD1 genes (See supplemental table).
Discussion
Loss of ARID1A expression is frequent in many types of cancers. Previous studies [21, 37] showed that almost 60% breast invasive ductal carcinoma exhibit loss of ARID1A expression and is associated with the most aggressive phenotypes of breast cancer. A study also showed that the lost expression of ARID1A was common in 30% of gastric cancer patients and was associated with poor clinical prognosis [38]. Current evidence on the importance of ARID1A shows that this gene acts as a tumor suppressor, and plays a key role in the progression of many types of cancer [13]. However, there are few studies that investigated the incidence and clinicopathologic importance of ARID1A loss in colorectal cancer and available published results are not conclusive [21, 23, 39]. For instance, Wei et al. showed that loss of ARID1A expression was associated with distant metastasis and late TNM stage of CRC; however, they did not find any association between loss of ARID1A expression and age, gender, tumor location and tumor size [21]. Whereas Chou et al. found that ARID1A loss was significantly associated with age, gender, tumor location and tumor size; however, they reported no association between ARID1A loss of expression and distant metastasis and TNM stage of CRC [23]. These contradictory results led us to investigate the incidence of ARID1A expression loss in CRC patients and analyze any association with clinicopathological parameters. Interestingly, our IHC results revealed that ARID1A was frequently reduced in CRC tissues when compared with paired normal tissues. Our results showed no ARID1A protein expression in 38.8% (7/18) cases of colorectal cancer and 66.6% had no or low expression. This suggests that ARID1A expression loss contributes to oncogenic transformation. Although we did not find any significant correlation between loss of ARID1A expression and both clinicopathological parameters and survival rates, the 66.6% incidence of negative and low ARID1A expression suggests that it is an important driver event in a large proportion of CRCs. In line with our results, Wei et al. observed that 25.8% of colorectal cancer tumors had loss of ARID1A expression and 51.2% of the tumors had low ARID1A expression compared with adjacent non-malignant tissue samples [21]. Overall, current IHC results and some previous studies [21, 23, 39, 40] suggest that ARID1A loss is not rare in colorectal cancer tumors. However, the molecular basis of this downregulation remains to be elucidated.
With regard to the clinicopathologic correlates and ARID1A expression loss, our investigation revealed no significant association between the loss of ARID1A expression and any clinicopathological parameters examined, including MSI status. Our results are in line with the study of Lee et al. [39]. These authors reported that ARID1A expression loss was not related to MSI status, TNM stage, pathologic differentiation, tumor location and tumor size. Similar findings were also reported by Rahman et al. [41], who reported no significant association between ARID1A loss and common clinicopathological parameters in uterine endometrioid carcinomas. In this context, it is also worth noting that in a study by Katagiri et al. using cervical adenocarcinomas showed high frequency of ARID1A expression loss but reported no significant association between the loss of ARID1A expression and any clinicopathological parameters. These authors speculated that one possible explanation for lack of correlation is that loss of ARID1A expression is an early event in the development of cancer. ARID1A protein might act as a “brake” by preventing excessive cellular proliferation and is essential for normal cell cycle arrest. This suggests that loss of ARID1A protein expression might not be as critical to tumor progression as to tumor initiation [42]. Notably, inconsistent reports also exist [21, 23, 40, 43]. Collectively, with respect to clinicopathologic importance of ARID1A loss, the documented results to date are controversial. These discrepancies might be attributed to differences in sample sizes, tissue- and organ-specific tumorigenesis pathways and different types of used IHC antibodies (mono vs. polyclonal).
Our study showed no significant association between overall survival and loss of ARID1A expression in CRC. However, patients with negative ARID1A expression had a tendency towards better overall survival than those with positive ARID1A expression. Our results are consistent with previous reports on CRC [23, 39, 40]. The previous studies did not find any significant association between loss of ARID1A expression and overall survival; however, they observed that overall survival was better for patients with negative ARID1A expression than those with positive ARID1A expression [23, 39, 40]. Only one previous study found a significant association between loss of ARID1A expression and overall survival in stage IV colorectal cancer, rather than in stage I-III patients. However, the small number of patients examined in the current study is not sufficient to determine the prognostic impact of ARID1A expression in CRC. Hence, further studies are required to clarify the prognostic value of ARID1A in CRC.
In order to characterize molecular mechanisms affecting ARID1A expression, we evaluated the possible role of promoter methylation in the downregulation of the ARID1A expression in various types of colorectal cell lines. A study showed that ARID1A promoter methylation led to decreased ARID1A expression in breast cancer [17]. However, there has been no investigation on ARID1A epigenetic inactivation in colorectal cancer. The present study evaluated ARID1A promoter methylation, and its association with the ARID1A expression levels in human CRC cell lines. ARID1A mRNA level was different in colorectal cancer cell lines and varied from almost nondetectable in SW48 to high expression level in HCT116 and HT-29/219. Hypermethylation was further detected in 4/6 (66%) CRC cell lines. This study showed that the SW48 cell line, displaying the ARID1A methylated allele only, did not express detectable ARID1A. In contrast, HCT116 and HT-29/219 displayed an unmethylated allele and a methylated allele, which was not detectable. We have also shown that treatment with the 5-aza dramatically restored ARIA1A expression in low ARID1A expressing cell lines, SW48 and SW742.
To sum up, the present study identified that promoter DNA hypermethylation strongly contributes to ARID1A silencing or downregulation in the CRC cell lines. However, other processes such as histone modification cannot be excluded. Indeed, ARID1A expression was not restored after 5-aza treatment in SW480 and LS180 cell lines. Zhang et al. carried out chromatin immunoprecipitation assays to evaluate ARID1A promoter histone modification levels in breast cancer samples and reported methylation of histone H3 at lysine 27 was linked to ARID1A silencing [17]. These findings suggest that different regulatory processes might play a role in ARID1A expression in different CRC lines. ARID1A is regulated by DNA methylation and repressive histone modifications and as such likely acts as a potent tumor suppressor gene in some subtypes of colorectal tumors, as it affects many genes’ expression through its role in chromatin remodeling.
As previously mentioned, a significant association between loss of ARID1A expression and MSI because of hypermethylation of the MLH1 gene promoter was reported in various cancers, including CRC [26, 40, 43]. It was speculated that ARID1A expression loss would result in aberrant epigenetic alterations by a deficient SWI/SNF complex with subsequent promoter hypermethylation of the MLH1 gene. Alternatively hypermethylation of the ARID1A promoter could be considered as an inactivating mechanism of ARID1A expression, and the co-occurrence of loss of ARID1A expression and MLH1 promoter hypermethylation might be the consequently a reflection of global genomic hypermethylation [26, 43]. This last hypothesis might be applicable to colorectal tumors, as we identified promoter hypermethylation as a silencing or downregulation mechanism of the ARID1A gene in the CRC cell lines. On a different note, lymphatic penetration was more pronounced in no/low ARID1A expression cases when compared to moderate/high expressing cases (33% vs. 16.66%). This finding suggests that lack of ARID1A expression renders the tumors more aggressive and invasive to neighboring tissues. Indeed, Kishida et al. have reported that negative ARID1A expression was correlated with early onset, lymphatic invasion, and lymph nodes metastasis and was also associated with specific mutational profile in PKHD1, RNF213, and MSH6 genes whereas KRAS mutations were more common in ARID1A-positive tumors [44].
Previous studies showed that copy number loss in ARID1A was the major mechanism for loss of expression in pancreatic cancer [28]. In addition, several previous studies indicated copy number loss occurs in 13–35% in breast cancers [17]. These led us to investigate whether copy number loss is also involved in the low ARID1A expression in the CRC cell lines. The comparison between ARID1A copy number in the CRC cell lines and normal sample revealed that CRC cell lines had no significant difference in ARID1A copy number and values were similar to the control sample (Fig. 3). These data suggest that the CNV is not for a player in the observed differences in ARID1A expression in the CRC cell lines.
This study has several limitations. The possible significance of ARID1A promoter hypermethylation in CRC requires further elucidation including complete CpG island promoter analysis that will be our goal for future studies using previous protocols [45]. In addition, the correlation between the ARID1A expression and promoter methylation requires to be assessed in patients with CRC and in a greater number of CRC cell lines. We are aware of other epigenetic mechanisms of silencing such as aberrant methylation of an upstream regulatory gene, dysregulation of miRNA expression, mutations and deletion that can possibly play a role in ARID1A expression in CRC.
In the case of our finding that loss of ARID1A expression was not correlated to survival and clinicopathological parameters, the limited number of patients might have limited our analysis. Further investigations with a larger number of patients are warranted to establish clinical and pathological impact of ARID1A expression loss in CRC.
Conclusion
We found a high incidence of ARID1A expression loss in CRC that suggests ARID1A as a tumor suppressor. There was no significant association between the loss of ARID1A expression and any of the clinicopathological parameters examined. Here we identified aberrant methylation of ARID1A gene as an important regulator of ARID1A expression in CRC cell lines and clinical specimens. The clinical significance of loss of ARID1A in CRC remains a goal for future studies.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- ARID1A:
-
AT-rich interactive domain-containing protein 1A
- CRC:
-
colorectal cancer
- MMR:
-
Mismatch repair
- EMAST:
-
Elevated microsatellite alterations at selected tetranucleotide repeats
- MSI:
-
Microsatellite Instability
- CNV:
-
Copy number variation
- qPCR:
-
Real-time quantitative PCR
References
Ashktorab H, Kupfer SS, Brim H, Carethers JM. Racial disparity in gastrointestinal cancer risk. Gastroenterology. 2017;153(4):910–23.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Fearon ER. Molecular genetics of colorectal cancer. Ann Rev Pathol Mech Dis. 2011;6:479–507.
Siegel R, Naishadham D, Jemal A. Cancer statistics for hispanics/latinos, 2012. CA Cancer J Clin. 2012;62(5):283–98.
Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8(12):686.
Dallas PB, Cheney IW, Liao D-W, Bowrin V, Byam W, Pacchione S, Kobayashi R, Yaciuk P, Moran E. p300/CREB binding protein-related protein p270 is a component of mammalian SWI/SNF complexes. Mol Cell Biol. 1998;18(6):3596–603.
Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol. 2013;43(9):849–55.
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–43.
Mitelman F, Johansson B, Mandahl N, Mertens F. Clinical significance of cytogenetic findings in solid tumors. Cancer Genet Cytogenet. 1997;95(1):1–8.
Guo XQ, Zhang QX, Huang WR, Duan XL, Cai ZM. Tumor suppressor role of chromatin-remodeling factor ARID1A. Yi Chuan. 2013;35(3):255–61.
Birnbaum DJ, Adélaïde J, Mamessier E, Finetti P, Lagarde A, Monges G, Viret F, Gonçalvès A, Turrini O, Delpero JR. Genome profiling of pancreatic adenocarcinoma. Genes Chromosom Cancer. 2011;50(6):456–65.
Guan B, Wang T-L, Shih I-M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71(21):6718–27.
Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33(1):100–3.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481.
Weissman B, Knudsen KE. Hijacking the chromatin remodeling machinery: impact of SWI/SNF perturbations in cancer. Cancer Res. 2009;69(21):8223–30.
Wang X, Nagl NG, Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112(4):636–42.
Zhang X, Sun Q, Shan M, Niu M, Liu T, Xia B, Liang X, Wei W, Sun S, Zhang Y. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLoS One. 2013;8(1):e53931.
Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci. 2008;105(51):20380–5.
Nagl NG, Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007;26(3):752–63.
Nagl NG, Patsialou A, Haines DS, Dallas PB, Beck GR, Moran E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005;65(20):9236–44.
Wei X-L, Wang D-S, Xi S-Y, Wu W-J, Chen D-L, Zeng Z-L, Wang R-Y, Huang Y-X, Jin Y, Wang F. Clinicopathologic and prognostic relevance of ARID1A protein loss in colorectal cancer. World J Gastroenterol: WJG. 2014;20(48):18404.
Somsuan K, Peerapen P, Boonmark W, Plumworasawat S, Samol R, Sakulsak N, Thongboonkerd V. ARID1A knockdown triggers epithelial-mesenchymal transition and carcinogenesis features of renal cells: role in renal cell carcinoma. FASEB J. 2019;33(11):12226–39.
Chou A, Toon CW, Clarkson A, Sioson L, Houang M, Watson N, DeSilva K, Gill AJ. Loss of ARID1A expression in colorectal carcinoma is strongly associated with mismatch repair deficiency. Hum Pathol. 2014;45(8):1697–703.
Guan B, Mao T-L, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, Chen E, Jeng Y-M, Wang T-L, Shih I-M. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011;35(5):625.
Kim MS, Je EM, Yoo NJ, Lee SH. Loss of ARID1A expression is uncommon in gastric, colorectal, and prostate cancers. Apmis. 2012;120(12):1020–2.
Bosse T, Ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, Nout RA, Creutzberg CL, Morreau H, Smit VT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26(11):1525.
Putra J, Suriawinata AA. Clinical significance of loss of ARID1A expression in colorectal and small intestinal carcinoma. Clin Transl Gastroenterol. 2015;6(12):e131.
Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, Maitra A, Pollack JR. Convergent structural alterations define SWItch/sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci. 2012;109(5):E252–9.
Wu J, Roberts C. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. https://doi.org/10.1158/2159-8290 In.: CD-12-0361.[PMC free article][PubMed] [Cross Ref].
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330.
Cajuso T, Hänninen UA, Kondelin J, Gylfe AE, Tanskanen T, Katainen R, Pitkänen E, Ristolainen H, Kaasinen E, Taipale M. Exome sequencing reveals frequent inactivating mutations in ARID1A, ARID1B, ARID2 and ARID4A in microsatellite unstable colorectal cancer. Int J Cancer. 2014;135(3):611–23.
Ashktorab H, Mokarram P, Azimi H, Olumi H, Varma S, Nickerson ML, Brim H. Targeted exome sequencing reveals distinct pathogenic variants in Iranians with colorectal cancer. Oncotarget. 2017;8(5):7852.
Brim H, Abu-Asab MS, Nouraie M, Salazar J, DeLeo J, Razjouyan H, Mokarram P, Schaffer AA, Naghibhossaini F, Ashktorab H. An integrative CGH, MSI and candidate genes methylation analysis of colorectal tumors. PLoS One. 2014;9(1):e82185.
Torshizi Esfahani A, Seyedna SY, Nazemalhosseini Mojarad E, Majd A, Asadzadeh Aghdaei H. MSI-L/EMAST is a predictive biomarker for metastasis in colorectal cancer patients. J Cell Physiol. 2019;234(8):13128–36.
Munakata K, Koi M, Kitajima T, Tseng-Rogenski SS, Uemura M, Matsuno H, Kawai K, Sekido Y, Mizushima T, Toiyama Y, Yamada T, Mano M, Mita E, Kusunoki M, Mori M, Carethers JM. Inflammation-associated microsatellite alterations caused by MSH3 dysfunction are prevalent in ulcerative colitis and increase with neoplastic advancement. Clin Transl Gastroenterol. 2019;10:e00105.
Mokarram P, Naghibalhossaini F, Firoozi MS, Hosseini SV, Izadpanah A, Salahi H, Malek-Hosseini SA, Talei A, Mojallal M. Methylenetetrahydrofolate reductase C677T genotype affects promoter methylation of tumor-specific genes in sporadic colorectal cancer through an interaction with folate/vitamin B12 status. World J Gastroenterol: WJG. 2008;14(23):3662.
Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, Edgren H, Kallioniemi O, Aleynikova O, Przybytkowski E. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene. 2012;31(16):2090.
Yan H-B, Wang X-F, Zhang Q, Tang Z-Q, Jiang Y-H, Fan H-Z, Sun Y-H, Yang P-Y, Liu F. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2013;35(4):867–76.
Lee SY, Kim D-W, Lee HS, Ihn MH, Oh H-K, Park DJ, Kim H-H, Kang S-B. Loss of AT-rich interactive domain 1A expression in gastrointestinal malignancies. Oncology. 2015;88(4):234–40.
Lee LH, Sadot E, Ivelja S, Vakiani E, Hechtman JF, Sevinsky CJ, Klimstra DS, Ginty F, Shia J. ARID1A expression in early stage colorectal adenocarcinoma: an exploration of its prognostic significance. Hum Pathol. 2016;53:97–104.
Rahman M, Nakayama K, Rahman MT, Katagiri H, Katagiri A, Ishibashi T, Ishikawa M, Iida K, Miyazaki K. Clinicopathologic analysis of loss of AT-rich interactive domain 1A expression in endometrial cancer. Hum Pathol. 2013;44(1):103–9.
Katagiri A, Nakayama K, Rahman MT, Rahman M, Katagiri H, Ishikawa M, Ishibashi T, Iida K, Otsuki Y, Nakayama S. Frequent loss of tumor suppressor ARID1A protein expression in adenocarcinomas/adenosquamous carcinomas of the uterine cervix. Int J Gynecol Cancer. 2012;22(2):208–12.
Ye J, Zhou Y, Weiser MR, Gönen M, Zhang L, Samdani T, Bacares R, DeLair D, Ivelja S, Vakiani E. Immunohistochemical detection of ARID1A in colorectal carcinoma: loss of staining is associated with sporadic microsatellite unstable tumors with medullary histology and high TNM stage. Hum Pathol. 2014;45(12):2430–6.
Kishida Y, Oishi T, Sugino T, Shiomi A, Urakami K, Kusuhara M, Yamaguchi K, Kitagawa Y, Ono H. Associations between loss of ARID1A expression and Clinicopathologic and genetic variables in T1 early colorectal Cancer. Am J Clin Pathol. 2019;152(4):463–70.
Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33(18):5868–77.
Acknowledgements
This project was part of the PhD thesis written by Mehran Erfani.
Funding
This study was supported (in part by a grant # 94–9136) from Shiraz University Medical Sciences, and (in part) by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number G12MD007597 and NIH CA206010. A. Alfred Taubman Medical Institute of the University of Michigan.
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: PM, ME, HA, JMC; performed experiments: ME, PM, HA, HB, MK; analyzed data: ME, AT, PM, HA, HB, MN, MZ,MT, MA, MK, SVH, and KT; contributed reagents/materials/analysis tools: EM, PM, HA, HB, MZ, MT, MA, JMC, MK, and KT; wrote and edited manuscript: ER, PM, HA, HB, and JSC; provided statistical analysis. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This retrospective and chart review study was conducted according to the World Medical Association Declaration of Helsinki and was approved by the Internal Review Board of Howard University and Shiraz Medical Science University. Since the chart review and collection of archival tissue was done through un-identifiable approach no consent form needed for this study. None of the cell lines used in the present study required ethics approval for their use.
Consent for publication
Not applicable.
Competing interests
Hassan Ashktorab and Hassan Brim do not have any conflict of interest for this research.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Erfani, M., Hosseini, S.V., Mokhtari, M. et al. Altered ARID1A expression in colorectal cancer. BMC Cancer 20, 350 (2020). https://doi.org/10.1186/s12885-020-6706-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-6706-x