
Abstract
Background
Poly (ADP-ribose) polymerase inhibitors targeting BRCA1/2 mutations are available for treating patients with high-grade serous ovarian cancer. These treatments may be more appropriately directed to patients who might respond if the tumor tissue is additionally tested by next-generation sequencing with a multi-gene panel and Sanger sequencing of a blood sample. In this study, we compared the results obtained using the next-generation sequencing multi-gene panel to a known germline BRCA1/2 mutational state determined by conventional Sanger sequencing to evaluate the landscape of somatic mutations in high-grade serous ovarian cancer tumors.
Methods
Cancer tissue from 98 patients with high-grade serous ovarian cancer who underwent Sanger sequencing for germline BRCA1/2 analysis were consecutively analyzed for somatic mutations using a next-generation sequencing 170-gene panel.
Results
Twenty-four patients (24.5%) showed overall BRCA1/2 mutations. Seven patients (7.1%) contained only somatic BRCA1/2 mutations with wild-type germline BRCA1/2, indicating acquired mutation of BRCA1/2. Three patients (3.1%) showed reversion of germline BRCA1 mutations. Among the 14 patients (14.3%) with both germline and somatic mutations in BRCA1/2, two patients showed different variations of BRCA1/2 mutations. The next-generation sequencing panel test for somatic mutation detected other pathogenic variations including RAD51D and ARID1A, which are possible targets of poly (ADP-ribose) polymerase inhibitors. Compared to conventional Sanger sequencing alone, next-generation sequencing-based tissue analysis increased the number of candidates for poly (ADP-ribose) polymerase inhibitor treatment from 17.3% (17/98) to 26.5% (26/98).
Conclusions
Somatic mutation analysis by next-generation sequencing, in addition to germline BRCA1/2 mutation analysis, should become the standard of care for managing women with high-grade serous ovarian cancer to widen the indication of poly (ADP-ribose) polymerase inhibitors.
Similar content being viewed by others


Background
BRCA1/2 mutational loss of function is a primary driver of epithelial ovarian cancer and is the basis of therapeutics targeting a synthetic lethality mechanism of poly (ADP-ribose) polymerase (PARP) inhibition in combination with BRCA1/2 mutation or possibly other homologous recombination genetic deficiencies [1, 2].
Most patients evaluated in previous PARP inhibitor-related randomized trials showed germline BRCA1/2 mutations [3]. However, the results of these studies may also be applicable to patients with somatic BRCA1/2 mutations [4, 5]. In 2014, the PARP inhibitor olaparib (Lynparza™, AstraZeneca, Cambridge, UK) was approved for treating patients with relapsed ovarian cancer with germline BRCA1/2 mutations by the US Food and Drug Administration and European Medicines Agency and for patients with somatic BRCA1/2 mutations by the European Medicines Agency [6].
In high-grade serous ovarian cancer (HGSOC), which comprises the majority of epithelial ovarian cancer cases, germline and somatic mutations in BRCA1/2 are detected in 17–25% of patients, with somatic mutations representing 18–30% of all BRCA1/2 mutations [7,8,9]. Analysis of ovarian cancer tissue from patients with HGSOC showed that loss of the normal copy of BRCA1/2 occurs in most germline BRCA1/2 mutations, indicating that this is an early event in HGSOC development [10].
In this study, we performed (i) next-generation sequencing (NGS) to determine the mutational state of BRCA1/2 in ovarian cancer tissues from 98 consecutive patients with HGSOC; (ii) compared the results to the known germline BRCA1/2 mutational state by conventional Sanger sequencing of blood samples; and (iii) determined the genetic landscape of somatic mutations in HGSOC tumors.
Methods
Study population
An electronic medical record review of patients treated for HGSOC at the Department of Obstetrics and Gynecology at the Severance Hospital of Yonsei University between January 2017 and February 2019 was carried out. Ninety-eight patients with HGSOC who were tested for both germline and somatic BRCA1/2 mutations were included in the analysis. The medical record and pedigree of each patient were reviewed, and data including age at HGSOC diagnosis, family history of BRCA1/2-related cancer, history of primary breast cancer, residual disease after cytoreductive surgery, and survival status were collected. A patient was considered to have a family history of BRCA1/2-related cancer if there were one or more instances of ovarian, peritoneal, fallopian tube, breast, pancreas, or prostate cancer among first- or second-degree relatives. This study was reviewed and approved by our organization’s institutional review board and was performed in accordance with the ethical standards described in the Declaration of Helsinki.
Genetic testing for germline BRCA1/2 mutations
All patients underwent in-house testing as previously reported [9]. Briefly, we identified all small base pair variations by Sanger sequencing on a 3730 DNA Analyzer with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Sequencing data were aligned against appropriate reference sequences and analyzed using Sequencher 5.3 software (Gene Codes Corp., Ann Arbor, MI, USA).
Genetic testing for somatic mutations using NGS multi-gene panel
All the tissue used for the NGS analysis was obtained at the first time of taking cancer tissue, i.e. at the time of primary debulking surgery or diagnostic laparoscopy. Formalin-fixed paraffin-embedded (FFPE) sections (5-μm-thick) were deparaffinized in xylene, hydrated through graded alcohols to water, and stained with Gill’s hematoxylin. The slides were manually microdissected under a dissecting microscope using a scalpel point dipped in ethanol. The scraped material was washed in phosphate-buffered saline and digested in proteinase K overnight at 37 °C in ATL Buffer (Qiagen, Hilden, Germany). DNA and RNA were then isolated using the QIAamp DSP DNA FFPE extraction kit (cat # 60404) and RNeasy FFPE kit (cat # 73504) according to the manufacturer instructions.
Mutational and copy number analysis was carried out using the Illumina TST-170 panel, according to the manufacturer instructions (San Diego, CA, USA). The gene panels cover 170 cancer-related genes for mutational analysis and 59 genes for copy number analysis (Supplementary Table 1). For mutational analysis, FASTQ files were uploaded on Illumina’s BaseSpace software for variant interpretation. Only variants in coding regions and promoter regions or splice variants were retained. In addition, only variants present in < 1% of the population according to ExAC and 1000 Genomes databases, and which were present in > 5% of reads with a minimum read depth of 250, were retained. All retained variants were reviewed using reference websites [Catalogue of Somatic Mutations in Cancer (http://evs.gs.washington.edu/EVS/), Precision Oncology Knowledge Base (http://oncokb.org), and dbSNP (https://www.ncbi.nlm.nih.gov/snp)], and only pathogenic variants were selected. In copy number analysis, genes showing greater than 2-fold change compared to the average level were considered to have undergone amplification. Genes showing a lower than 0.7-fold change compared to the average levels were considered to exhibit significant copy number loss. Fusion and splice variants were detected by RNA analysis workflow in the TST-170 panel. RNA was converted to cDNA in the first step, and the remaining steps of NGS library preparation, hybrid-capture based enrichment, and sequencing were similar to the workflow of the DNA analysis module of TST-170 except for the hybrid-capture probes for 55 genes included in the RNA analysis workflow. Data analysis for fusion and splice variants was performed with TST-170 Local App provided by Illumina. Specifically, Manta was used for the fusion variant calling. For splice variant calling, Illumina’s RNA Splice Variant Calling software was used.
Statistical analyses
IBM SPSS version 23 for Windows (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. The Kolmogorov–Smirnov test was used to validate standard normal distribution assumptions. Pearson’s chi-square test, Fisher’s exact test, Student’s t-test and Mann–Whitney U-test were used for univariate analysis. Survival outcomes were determined using Kaplan–Meier survival analysis.
Results
Study population
Patient characteristics are shown in Table 1. Of the 98 patients, 46 patients received neoadjuvant chemotherapy (NAC) after diagnostic laparoscopy. There was no difference in the proportion of patients who treated with NAC between the overall BRCA1/2 mutation group and the BRCA1/2 wild-type group. All the patients receive platinum-based chemotherapy, and PARP inhibitor was not used. Patients with overall BRCA1/2 mutations tended to show a higher rate of BRCA1/2-related family history and breast cancer history compared to patients with wild-type BRCA1/2. However, no factors showed a significant difference. Overall BRCA1/2 mutation appeared to be correlated with a better prognosis than wild-type BRCA1/2, which was comparable to the results of our previous study; however, this tendency was not significant, likely because of the small number of patients (Fig. 1) [11].

Comparison of survival between overall BRCA mutation and BRCA wild-type in the Kaplan-Meier curve. BRCAm, BRCA mutation; BRCAw, BRCA wild-type
Frequency and spectrum of germline and somatic BRCA1/2 mutation
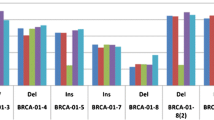
Figure 2 shows the distribution of germline and somatic BRCA1/2 mutations in this population. Twenty-four (24.5%) of the 98 patients had either germline or somatic BRCA1/2 mutations. Among the 24 patients with mutations in BRCA1 or BRCA2, 14 showed both germline and somatic mutations. However, three and seven patients contained only germline and only somatic mutations, indicating reversion (Reversion #1–3) and acquired mutation (Acquired #1–#7) of BRCA1/2, respectively (Table 2). Interestingly, even among the 14 patients who had both germline and somatic mutations, two showed variations in BRCA1/2 (Replace #1–#2). The inconsistent BRCA1/2 status is presented in Table 2.

Distribution of germline and somatic BRCA1/2 mutations. Pts, patients
Landscape of somatic mutations shown in NGS multi-gene panel
The landscape of somatic mutations of the included patients is shown in Fig. 3. Ten mutated genes were detected in 92 (93.9%) of the 98 patients: TP53, BRCA2, BRCA1, KRAS, ARID1A, RB1, PIK3CA, STK11, FGFR2, and RAD51D. Among them, four genes, TP53, BRCA1, BRCA2, and KRAS, were detected in multiple patients. TP53 mutation was observed in 90 (91.8%) patients, including missense mutation (52, 57.8%), frameshift (19, 21.1%), nonsense mutation (16, 17.8%), and in-frame deletion (3, 3.3%). BRCA1 and BRCA2 mutations were detected in 11 (11.2%) and 12 (12.2%) patients, respectively. All patients who had BRCA1 or BRCA2 mutations showed TP53 mutation. KRAS mutation was detected in 5 patients (5.1%). Additionally, BRCA1/2, ARID1A, and RAD51D mutations, which are introduced by homologous recombination, may be targets of PARP inhibitors [12, 13]. The median depth of NGS sequencing was 880.5 (range, 280–1337). All information on patients enrolled in this study is presented in Supplementary Table 2.

Landscape of somatic mutations and germline BRCA1/2 mutations in 98 patients with HGSOC. HGSOC, high-grade serous ovarian cancer
Somatic mutations were compared to those in 316 patients with serous ovarian cancer from The Cancer Genome Atlas (Supplement Figure 1). TP53 mutation was detected in 88% of patients (277 patients), which was comparable to the value in our data. However, BRCA1 and BRCA2 were observed in 4% (12 patients) and 3% of patients (11 patients); these values were considerably smaller than those in our data.
Discussion
Principal findings
In this study, we compared the germline and somatic BRCA1/2 mutation status of 98 patients with HGSOC. Twenty-four (24.5%) of the 98 patients had either germline or somatic BRCA1/2 mutations. Three of 17 patients (17.6%) showed restored BRCA1/2 mutation, and seven of 81 patients (8.6%) exhibited acquired BRCA1/2 mutations. These data indicate that among the patients who were negative for germline BRCA1/2 mutation, approximately 10% may have only somatic BRCA mutations without germline mutation.
Results
NGS-based multi-gene panel testing of ovarian cancer tissue allows for the identification of somatic mutations that are not detected by blood-based examination and are often not identified at low allele frequencies by Sanger sequencing of tumor samples. The tumor assay used in this study showed that 93.9% (92/98) of patients with somatically mutated tumors, including TP53, BRCA1, BRCA2, KRAS, ARID1A, RB1, PIK3CA, STK11, FGR2, and RAD51D, were not adequately detected by Sanger sequencing and BRCA1/2 testing of clinical FFPE sections. Furthermore, the ability to determine the mutational status of 170 cancer genes simultaneously provides insight into the co-occurrence patterns of mutations, additional oncogenic drivers, and intra- or inter-tumor heterogeneity, and is useful for identifying homologous recombination and DNA repair genes beyond BRCA1/2 which may be involved in the response to PARP inhibitors. ARID1A and RAD51D mutations were found in 2 patients, demonstrating that these patients are possible candidates for PARP inhibitor treatment [12, 13]. Thus, compared to conventional Sanger sequencing alone, NGS-based tissue analysis increased the number of candidates for PARP inhibitor treatment from 17.3% (17/98) to 26.5% (26/98).
TP53 mutation was found in 90 (91.8%) patients. The overall frequency of TP53 mutation in the 316 patients from The Cancer Genome Atlas 2011 study was 88.0% (277/316), which is comparable to our results (Supplement Figure 1). All patients with BRCA1/2 mutation showed TP53 mutation, indicating that BRCA1/2 mutation is an earlier event than TP53 mutation. The correlation between BRCA1/2 and TP53 observed in this study is consistent with the results of previous studies evaluating patients with breast cancer [14].
Clinical implications
In the previous study, patients with ovarian cancer who did not carry germline BRCA1/2 mutations also responded to PARP inhibitors, suggesting that the broader dysfunction of genes, such as a homologous recombination-deficient phenotype, is important [15]. As initially reported in a previous phase II study of olaparib, objective responses were confirmed in 41% (7/17) of patients with ovarian cancer with germline BRCA mutations and 24% (11/46) of patients without germline mutations [16]. Remarkably, in responders belonging to the latter group, BRCA1/2 somatic mutations were detected. Subsequent studies of olaparib (Study 19) and rucaparib (ARIEL 2 and Study 10) confirmed that BRCA-mutated patients derived the most significant clinical benefit from PARP inhibitor treatment and showed no differences in responsiveness to PARP inhibitors between germline and somatic BRCA-mutated HGSOC [17,18,19]. Both BRCA1, located on chromosome 17 (17q21), and BRCA2, on chromosome 13 (13q12.3), are very large, and exon 11 of both is thought to encode relevant protein domains as mutations in these regions are highly pathogenic [10, 20]. However, because of the gene length and domain complexity, pathogenic mutations may occur anywhere and can be highly variable as well as depend on ethnicity [21, 22].
Since the introduction of PARP inhibitors as the first targeted therapy, ovarian cancer diagnosis has involved not only standard morphological and immunophenotypic evaluation of cancer samples, but also detailed genotyping and mutational profiling. Additionally, an understanding of the mutational status in genes essential for drug sensitivity and resistance is necessary to ensure effective treatment of ovarian cancer. As more studies are conducted and targeted therapeutics become available, genomic analysis for cancer diagnosis and treatment will benefit a large number of patients who currently have unmet medical needs.
Research implications
Further studies are required to determine whether the extent and duration of benefit in patients with germline and somatic BRCA1/2 mutations are equivalent. Additionally, the appropriate time for obtaining the tumor tissue for somatic mutational analysis must be determined. Specifically, whether previously archived FFPE sections miss patients whose somatic mutations are acquired later in their pathway of cancer should be evaluated. Notably, 100% of germline and 83% of somatic loss-of-function mutations showed biallelic inactivation and were predominantly clonal, suggesting that loss of function of BRCA can occur early in the development of HGSOC [23]. This finding indicates that retesting for somatic BRCA1/2 mutations using fresh biopsies at each relapse may not be informative, although data from ARIEL 2 showed controversial results regarding this point [19].
Low-grade serous ovarian cancer (LGSOC) was known to have a high prevalence of KRAS and BRAF mutations, but a low prevalence of TP53 mutations [24]. In our study, five cases showed KRAS mutation. However, three out of the five patients had simultaneous TP53 mutations as well. Additionally, contrary to the previous report, all the KRAS-mutated patients were diagnosed with HGSOC, not LGSOC. We hope to be able to report a further study that investigates the impact of the results of NGS panel on the conventional histologic diagnosis reversely, showing how much proportion of the conventional histologic diagnosis would be changed reflecting the results of NGS.
Strengths and limitations
This is the first study to compare the germline and somatic BRCA1/2 mutation status in patients of Asian ethnicity, which may guide future research of Asian patients with HGSOC. The current study had some limitations; it included a small number of patients and was performed in a single center. Additionally, NGS-based tests were not conducted as prospective schedules. Therefore, the timing of tests was variable among patients and the exact timing of the acquired or reversion of BRCA1/2 mutations could not be investigated. Also, technical challenges in identifying the mutations in tumors, such as difficulty in detecting mutation from archival tumor specimen and issues related with intratumoral heterogeneity, might be criticized as limitations of our study. Additionally, we could not show whether the “replace” or “reversion” cases really have the function of replaced or reversed BRCA1/2 protein, respectively.
Conclusions
The effectiveness of PARP inhibitors likely extends beyond the treatment of germline BRCA1/2 mutations to include homologous recombination deficiency in patients with HGSOC. NGS-based somatic mutation analysis, as well as germline BRCA1/2 mutation analysis, should become the standard of care for managing women with ovarian cancer to widen the indication of PARP inhibitors.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- FFPE:
-
Formalin-fixed paraffin-embedded
- HGSOC:
-
High-grade serous ovarian cancer
- NAC:
-
Neoadjuvant chemotherapy
- NGS:
-
Next-generation sequencing
- PARP:
-
Poly (ADP-ribose) polymerase
References
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, et al. Maintenance Olaparib in patients with newly diagnosed advanced ovarian Cancer. N Engl J Med. 2018;379(26):2495–505.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian Cancer. N Engl J Med. 2016;375(22):2154–64.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England). 2017;390(10106):1949–61.
Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al. FDA approval summary: Olaparib Monotherapy in patients with deleterious Germline BRCA-mutated advanced ovarian Cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–61.
The Cancer Genome Atlas Research Network, Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–75.
Eoh KJ, Park JS, Park HS, Lee ST, Han J, Lee JY, Kim SW, Kim S, Kim YT, Nam EJ. BRCA1 and BRCA2 mutation predictions using the BRCAPRO and myriad models in Korean ovarian cancer patients. Gynecol Oncol. 2017;145(1):137–41.
Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, Abkevich V, Potter J, Pruss D, Glenn P, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–6.
Eoh KJ, Park HS, Park JS, Lee ST, Han J, Lee JY, Kim SW, Kim S, Kim YT, Nam EJ. Comparison of clinical outcomes of BRCA1/2 pathologic mutation, variants of unknown significance, or wild type epithelial ovarian Cancer patients. Cancer Res Treat. 2017;49(2):408–15.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, Kapoor P, Ju Z, Mo Q, Shih Ie M, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discovery. 2015;5(7):752–67.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discovery. 2017;7(9):984–98.
Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res. 2001;61(10):4092–7.
Tinker AV, Gelmon K. The role of PARP inhibitors in the treatment of ovarian carcinomas. Curr Pharm Des. 2012;18(25):3770–4.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, McNeish IA, Swisher EM, Ray-Coquard I, Bell-McGuinn K, Coleman RL, O'Malley DM, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from study 10 and ARIEL2. Gynecol Oncol. 2017;147(2):267–75.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O'Malley DM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87.
Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. Jama. 2015;313(13):1347–61.
Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–71.
Eoh KJ, Park HS, Park JS, Lee ST, Han JW, Lee JY, Kim S, Kim SW, Kim YT, Nam EJ. Distinct clinical courses of epithelial ovarian Cancer with mutations in BRCA1 5′ and 3′ exons. Anticancer Res. 2018;38(12):6947–53.
Dougherty BA, Lai Z, Hodgson DR, Orr MCM, Hawryluk M, Sun J, Yelensky R, Spencer SK, Robertson JD, Ho TW, et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget. 2017;8(27):43653–61.
Kaldawy A, Segev Y, Lavie O, Auslender R, Sopik V, Narod SA. Low-grade serous ovarian cancer: a review. Gynecol Oncol. 2016;143(2):433–8.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2014R1A1A1A05002926, 2017R1A2B4005503) and faculty research grant of Yonsei University College of Medicine (6-2018-0053).
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2014R1A1A1A05002926, 2017R1A2B4005503) and faculty research grant of Yonsei University College of Medicine (6-2018-0053).
Author information
Authors and Affiliations
Contributions
Conception and design of the study were performed by KJE, HMK, JYL, SK, SWK, YTK, and EJN. Data was collected KJE and HMK. Data were analyzed and interpreted by KJE, HMK, and EJN. Statistical analysis was conducted by KJE. The manuscript was prepared by KJE, HMK, and EJN. Patients were recruited by KJE, JYL, SK, SWK, YTK, and EJN. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol received Institutional Review Board approval of the Yonsei University College of Medicine and was performed in accordance with the ethical standards described in the Declaration of Helsinki. The requirement to obtain a written informed consent was waived by the Institutional Review Board of the Yonsei University College of Medicine because our study was retrospective research based on medical records, and this research presented no more than minimal risk of harm to subjects.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplement Table 1.
Table of 170 genes evaluated in the NGS multi-gene panel in this study. The gene panels cover 170 cancer-related genes for mutational analysis and 59 genes for copy number analysis.
Additional file 2: Supplement Table 2.
All information on patients enrolled in this study.
Additional file 3: Supplement Figure 1.
Landscape of somatic mutations detected in this study in the TCGA database (316 patients with serous ovarian cancer). Somatic mutations were compared to those in 316 patients with serous ovarian cancer from The Cancer Genome Atlas.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eoh, K.J., Kim, H.M., Lee, JY. et al. Mutation landscape of germline and somatic BRCA1/2 in patients with high-grade serous ovarian cancer. BMC Cancer 20, 204 (2020). https://doi.org/10.1186/s12885-020-6693-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-6693-y