
Abstract
Background
Breast cancer is the most common malignancy in women worldwide. Although the endocrine therapy that targets estrogen receptor α (ERα) signaling has been well established as an effective adjuvant treatment for patients with ERα-positive breast cancers, long-term exposure may eventually lead to the development of acquired resistance to the anti-estrogen drugs, such as fulvestrant and tamoxifen. A better understanding of the mechanisms underlying antiestrogen resistance and identification of the key molecules involved may help in overcoming antiestrogen resistance in breast cancer.
Methods
The whole-genome gene expression and DNA methylation profilings were performed using fulvestrant-resistant cell line 182R-6 and tamoxifen-resistant cell line TAMR-1 as a model system. In addition, qRT-PCR and Western blot analysis were performed to determine the levels of mRNA and protein molecules. MTT, apoptosis and cell cycle analyses were performed to examine the effect of either guanine nucleotide-binding protein beta-4 (GNB4) overexpression or knockdown on cell proliferation, apoptosis and cell cycle.
Results
Among 9 candidate genes, GNB4 was identified and validated by qRT-PCR as a potential target silenced by DNA methylation via DNA methyltransferase 3B (DNMT3B). We generated stable 182R-6 and TAMR-1 cell lines that are constantly expressing GNB4 and determined the effect of the ectopic GNB4 on cell proliferation, cell cycle, and apoptosis of the antiestrogen-resistant cells in response to either fulvestrant or tamoxifen. Ectopic expression of GNB4 in two antiestrogen resistant cell lines significantly promoted cell growth and shortened cell cycle in the presence of either fulvestrant or tamoxifen. The ectopic GNB4 induced apoptosis in 182R-6 cells, whereas it inhibited apoptosis in TAMR-1 cells. Many regulators controlling cell cycle and apoptosis were aberrantly expressed in two resistant cell lines in response to the enforced GNB4 expression, which may contribute to GNB4-mediated biologic and/or pathologic processes. Furthermore, knockdown of GNB4 decreased growth of both antiestrogen resistant and sensitive breast cancer cells.
Conclusion
GNB4 is important for growth of breast cancer cells and a potential target for treatment.
Similar content being viewed by others

Background
Breast cancer is the most common malignancy found in women worldwide and the second leading cause of cancer-related deaths among North American women [1]. Globally, it is estimated that 1.67 million new cases were diagnosed, and 522,000 women died from this disease in 2012 (GLOBOCAN 2012). Approximately 60% of these deaths were contributed by less developed countries. Although the exact etiology of breast cancer is currently unknown, the estrogen/estrogen-receptor (ER) signaling may play a crucial role in the development of this disease [2,3,4,5]. Furthermore, sustained estrogenic exposure may also increase the risk of breast, ovarian, and endometrial cancers [6].
Estrogen receptor alpha (ERα) and beta (ERβ), two ligand-inducible transcription factors, are members of the steroid/thyroid receptor superfamily that primarily mediate estrogen’s biological function through binding [7]. ERα and ERβ are encoded by either ESR1 or ESR2 genes that are composed of 595 and 530 amino acids, respectively. Both eventually form an N-terminal domain (NTD), a DNA-binding domain (DBD), and a ligand-binding domain (LBD) [8]. Sequence analysis indicates that ERα and ERβ display ~ 97% similarity in the DBD and 59% in the LBD, whereas they display only 16% similarity in the NTD [8]. This implicates a functional similarity and difference between these two ERs. For instance, once activated via estrogen binding, both dimerized ERs can either bind to the estrogen-response element (ERE) in the DNA or interplay with other transcription factors, such as AP1, Sp1, and NF-κB [9], eventually influencing the transcription of genes. However, ERα may predominantly bind to ERE elements [10], while ERβ may primarily interact with AP1 sites [11]. Furthermore, as demonstrated, ERα is a key player in promoting cell growth and proliferation [12, 13], whereas ERβ plays an important role in anti-proliferation, differentiation, and apoptosis in human malignancies, including breast cancer [14, 15].
Because ERα is expressed in 70% of breast cancers [16], and the proliferation of these ERα-positive breast cancers is largely dependent on estrogen/ERα signaling [17], the endocrine therapy that targets estrogen/ERα signaling has been well established as an effective adjuvant treatment for patients with ERα-positive breast cancers [18]. The endocrine-therapy agents that are currently used for ERα-positive breast cancer include fulvestrant (also known as ICI 182,780 and faslodex, the ER downregulator that selectively downregulates and/or degrades ERα), tamoxifen (the ER modulator that selectively antagonizes ERα function), and aromatase inhibitors (e.g. letrozole and anastrozole, which inhibit estrogen production by attenuating aromatase activity) [17, 19]. As an important adjuvant therapy, continuing 10-year tamoxifen treatment, when compared with 5-year exposure, has been shown to further reduce the risk of disease recurrence and mortality in a randomized trial of women with ER-positive breast cancers [20]. Unfortunately, long-term exposure may eventually lead to the development of acquired resistance to these drugs [21,22,23], which is a serious clinical problem in hormonal therapy. However, the underlying mechanisms are not completely understood.
In this study, we globally analyzed genomic DNA methylation, correlated with gene expression profiling, and identified GNB4 that was silenced by DNMT3B-mediated DNA methylation in both fulvestrant-resistant (MCF-7/182R-6) and tamoxifen-resistant (MCF-7/TAMR-1) breast cancer cell lines. Ectopic expression of GNB4 enhanced proliferation of MCF-7/182R-6 and MCF-7/TAMR-1 cell lines in response to either fulvestrant or tamoxifen, while it shortened G2 and S phases in the cell cycle. We also noted that the ectopic expression of GNB4 induced apoptosis in the MCF-7/182R-6 cell line, whereas it attenuated the induction of apoptosis in the MCF-7/TAMR-1 cell line. Cell-cycle and apoptosis regulators were aberrantly expressed in these cell lines in response to the ectopic GNB4 expression. In contrast, siRNA-mediated knockdown of GNB4 inhibited proliferation of two resistant cell lines in the presence of either fulvestrant or tamoxifen, and induced either S phase arrest or apoptosis. Our results provide novel insight into the role of GNB4 in the growth of both antiestrogen-resistant and sensitive breast cancer cells and may represent a target for treatment of breast cancer.
Methods
Cell culture
The MCF-7/S0.5 (S05), MCF-7/182R-6 (182R-6), and MCF-7/TAMR-1 (TAMR-1) cell sublines were developed by Dr. Anne Lykkesfeldt (Breast Cancer Group, Cell Death and Metabolism, Danish Cancer Society Research Center, DK-2100, Copenhagen, Denmark). ICI 182,780 (Faslodex, fulvestrant) and tamoxifen-resistant sublines, 182R-6 and TAMR-1, respectively, are derived from S05 as described elsewhere [24, 25]. These cell lines were cultured in a DMEM/F-12 medium with 2.5 mM L-Glutamine, without HEPES and phenol red (HyClone), and supplemented with 1% heat-inactivated fetal bovine serum (HyClone). Additionally, for 182R-6 and TAMR-1 sublines were regularly supplemented with 0.1 μM ICI 182,780 and 1 μM tamoxifen, respectively. Human mammary epithelial cells (HMEC) purchased from ThermoFisher Scientific (Cat# A10565) were cultured in a HuMEC basal serum-free medium (ThermoFisher Scientific) containing HuMEC supplement (ThermoFisher Scientific), 100 IU/mL penicillin, and 100 mg/mL streptomycin. All cell lines were incubated at 37 °C in a humidified atmosphere of 5% CO2.
Whole-genome gene expression profiling
Total RNA was isolated from S05, 182R-6, and TAMR-1 cells using an Illustra RNAspin mini kit according to the manufacturer’s instructions (GE Healthcare Life Sciences). Quantification, purity, and integrity of the RNA samples were measured using a NanoDrop 2000c spectrophotometer (Thermo Scientific) and an Agilent 2100 bioanalyzer (Santa Clara). RNA samples with RIN values of seven or higher were used for further analysis. Procedures for library preparation, hybridization, detection, BeadChip statistical analysis, and data processing have been described previously [19]. Heatmaps were generated by Dr. Yaroslav Ilnytskyy for genes that were differentially expressed between any of the groups (ANOVA type analysis with p.adjusted < 0.001) and for top 1000 most variable probes in DNA methylation.
Whole-genome DNA methylation profiling
DNA was extracted from cells using the DNeasy Blood and Tissue Kit (QIAGEN) and treated with DNase-free RNase (Sigma) according to the manufacturer’s protocols. The collected DNA was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer’s protocols. Methylation was measured using the Infinium assay on the Illumina platform. Data was collected from the > 27,000 probes represented on the HM27 microarray. These probes contain CpG dinucleotides from selected loci throughout the genome. All steps were carried out according to the manufacturer’s specifications and with Illumina-supplied reagents. Briefly, bisulfate-converted samples were amplified overnight, fragmented, and purified. The re-suspended samples were hybridized overnight to the microarray, which harboured millions of bead-bound 50-mer oligos. Each interrogated loci is represented by two bead types: a methylated type (“C” remains a “C”) and an unmethylated type (“C” become a “T”). Hybridized chips were washed to remove unbound and/or non-specific DNA fragments. The resulting oligo-sample hybrid was then extracted with a biotin-linked dideoxy cytosine and stained with streptavidin. The relative intensity of the unmethylated bead to the methylated bead for each allele provides a measure of relative methylation levels.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA, isolated from the indicated cell lines with TRIzol reagent (Invitrogen), was subjected to qRT-PCR using an iScript™ Select cDNA Synthesis kit and an SsoFastMT EvaGreen Supermix (Bio-Rad) with the primer set specifically to GNB4 (PrimePCR SYBR Green Assay, Bio-Rad) according to the manufacturer’s instructions. Glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) was used as the internal control to standardize and test the RNA integrity with a sequence for the forward primer, 5′-GAA GGC TGG GGC TCA TTT-3′, and for the reverse primer, 5’-CAG GAG GCA TTG CTG ATG AT-3′ [26]. All qRT-PCR experiments were performed in triplicate, the data was analyzed using the comparative Ct method, and the results are shown as a fold induction of mRNA.
Knockdown of DNMT3B
182R-6 and TAMR-1 cells grown to 80% confluency were transiently transfected with either 200 nM Dnmt3b siRNA (Santa Cruz Biotechnologies) or 200 nM AllStars Negative Control siRNA (QIAGEN) using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. Seventy-two hours after transfection, the total RNA was isolated and subjected to qRT-PCR analysis using a primer set specifically to GNB4 (Bio-Rad) according to the manufacturer’s instructions, and the whole cellular lysates were prepared and subjected to Western blot analysis using antibodies against DNMT3B and GNB4.
Generation of GNB4 expression construct and GNB4 stable-expression cell lines
The coding sequence of GNB4 was amplified by RT-PCR using total RNA isolated from HMEC. The PCR product was then cloned into a pGEM-T easy vector (Promega), released by digestion with EcoR I and BamH I, and subcloned into a pEGFP-C1 vector (CloneTech) to generate pEGFP-GNB4. Sequence identity was confirmed by automatic sequencing. Primers used here for amplifying the GNB4 coding sequence are as follows: GNB4-F (5′-GAG AAT TCT ATG AGC GAA CTG GAA C-3′) and GNB4-R (5′-GGG GAT CCA TTC CAG ATT CTA AG-3′).
182R-6 and TAMR-1 cells grown to 80% confluency were transfected with either pEGFP-GNB4 or pEGFP-C1 using Lipofectamine 3000 (Invitrogen). 24 h after transfection, G418 was added to final concentrations of 800 μg/mL and 400 μg/mL for 182R-6 and TAMR-1 cell lines, respectively, to kill the negative cells. The positive cells stably expressing either GFP or GFP-GNB4 were further selected with cell sorting (University of Calgary).
MTT assay
The MTT assay was performed as described previously [27]. Briefly, 3.0 × 103 182R-6 or TAMR-1 or S05 cells transiently transfected with either 30 nM GNB4 (Gβ4) siRNA (Santa Cruz Biotechnology) or 30 nM AllStars negative control siRNA (QIAGEN), or stably expressing either GFP or GFP-GNB4 were plated in 96-well plates. The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays were carried out using a Cell Proliferation Kit I (Roche Diagnostics GmbH) according to the manufacturer’s instructions. The spectrophotometric absorbance of samples was measured at 595 nm using a microtiter plate reader (FLUOstar Omega).
Cell cycle and apoptosis analyses
182R-6 or TAMR-1 cells stably expressing either GFP or GFP-GNB4 grown to 90% confluency were harvested for cell-cycle and apoptosis analyses that were performed with a BD FACSCanto™ II Flow Cytometer (BD Biosciences) using a GFP-Certified Nuclear-ID Red Cell Cycle Analysis Kit (Enzo) and an Annexin V-Cy3 Apoptosis Kit Plus (BioVision) according to the manufacturer’s instructions.
182R-6 or TAMR-1 cells transiently transfected with either 30 nM GNB4 siRNA (Santa Cruz Biotechnology) or 30 nM AllStars negative control siRNA (QIAGEN), 72 h (for TAMR-1 line) or 96 h (for 182R-6 line) after transfection, the cells were harvested for cell-cycle and apoptosis analyses that were performed with a BD FACSCanto™ II Flow Cytometer (BD Biosciences) using propidium iodide staining solution and FITC Annexin V Apoptosis Detection kit II (BD Biosciences) according to the manufacturer’s instructions.
Western blot analysis
The indicated cells grown to 90% confluency were rinsed twice with ice-cold PBS and scraped off the plate in a radioimmunoprecipitation assay buffer (RIPA). We electrophoresed 30–100 μg of protein per sample on 6% or 10% SDS-PAGE and electrophoretically transferred to a PVDF membrane (Amersham Hybond™-P, GE Healthcare) at 4 °C for 1.5 h. Blots were incubated for one hour with 5% nonfat dry milk to block nonspecific binding sites and then incubated with polyclonal/monoclonal antibodies against BAX (BCL2-associated X protein), BCL2 (B-cell CLL/Lymphoma 2), DNMT3A (DNA methyltransferase 3A), GNB4, pAKT1/2/3 (phosphorylated AKT1/2/3) (Santa Cruz Biotechnology) or AKT1 (v-AKT murine thymoma viral oncogene homolog 1), DNMT1 (DNA methyltransferase 1), DNMT3B, p21(Waf1/Cip1) (Abcam) or CDK2 (cyclin-dependent kinase 2), CDK6 (cyclin-dependent kinase 6), cyclin A2, cyclin D1, cyclin E1, ERK1/2 (extracellular signal-regulated kinase 1/2), MeCP2 (methyl-CpG-binding protein 2), and pERK1/2 (phosphorylated ERK1/2) (Cell Signaling Technology) at 4 °C overnight. Immunoreactivity was detected using a peroxidase-conjugated antibody and visualized by an ECL Plus Western Blotting Detection System (GE Healthcare). The blots were stripped before re-probing with antibody against actin (Santa Cruz Biotechnology).
Statistical analysis
The student’s t-test was used to determine the statistical significance between groups in GNB4 expression, cell growth, cell cycle, and apoptosis. p < 0.05 was considered significant.
Results
Epigenetic silencing of GNB4 via DNMT3B-mediated DNA methylation
To explore the contribution of DNA methylation to the development of the acquired resistance to endocrine therapy in breast cancer, using a fulvestrant-resistant 182R-6 cell line, a tamoxifen-resistant TAMR-1 cell line, and their parental line S05 as a model system, we performed whole-genome DNA methylation and gene-expression profilings. We identified 284 genes as common targets of DNA methylation in both 182R-6 and TAMR-1 cell lines (Fig. 1a and c). Differential expression in both antiestrogen-resistant cell lines was evident in 210 genes (Fig. 1b and d). We then correlated the expression of 210 genes with their DNA methylation status and identified nine downregulated genes, including annexin A6 (ANXA6), dual-specificity phosphatase 2 (DUSP2), ephrin B3 (EFNB3), guanine nucleotide-binding protein beta-4 (GNB4), methyltransferase-like 7A (METTL7A), p8 (also known as candidate of metastasis 1, COM1), protease serine 23 (PRSS23), S100 calcium-binding protein A4 (S100A4), and tripartite motif-containing 4 (TRIM4). Their promoters were hypermethylated in both 182R-6 and TAMR-1 cell lines (Table 1), while only GNB4 was validated to be downregulated in both cell lines by qRT-PCR and Western blot analyses (Fig. 2a). Interestingly, Western blot analysis showed that DNMT3B was upregulated, while MeCP2 was downregulated in both cell lines. This implicates a common role of DNMT3B in both cell lines in GNB4 promoter hypermethylation, although DNMT1 may also play a role in the TAMR-1 cell line (Fig. 2b). To test our hypothesis, DNMT3B was transiently knocked down using siRNA. Seventy-two hours after transfection, DNMT3B was downregulated by siRNA; as a result, GNB4 was upregulated at both mRNA and protein levels in 182R-6 and TAMR-1 cell lines (Fig. 2c and d); whereas, DNMT3B siRNA had no effect on the expression of both DNMT1 and DNMT3A (Fig. 2d). Our results suggest that GNB4 was epigenetically silenced in 182R-6 and TAMR-1 cell lines by DNA methylation via DNMT3B.

Whole-genome DNA methylation and gene expression analyses. a, Heatmap of differentially methylated genes. DNA extracted from S05, 182R-6, and TAMR-1 cells was treated with DNase-free RNase and bisulfite converted; DNA methylation assay, data collection, and analysis were performed as described in “Methods”. b, Heatmap of differentially expressed genes. Total RNA isolated from S05, 182R-6, and TAMR-1 cells was subjected to gene expression profiling; the detailed procedures for library preparation, hybridization, detection, BeadChip statistical analysis, and data processing have been described previously [19]. c and d, The number of differentially methylated genes (c) and differentially expressed genes (d) was presented with a Venn diagram

Silencing of GNB4 in 182R-6 and TAMR-1 cells via DNMT3B. a, Total RNA isolated from S05, 182R-6, and TAMR-1 cells was subjected to qRT-PCR using a primer set specific to GNB4. Whole cellular lysate was prepared from S05, 182R-6, and TAMR-1 cells, and Western blot analysis was performed using an antibody against GNB4. b, Whole cellular lysate was prepared from S05, 182R-6, and TAMR-1 cells, and Western blot analysis was performed using antibodies against DNMT1, DNMT3A, DNMT3B, and MeCP2. c, 182R-6 and TAMR-1 cells were transiently transfected with either 200 nM DNMT3B siRNA or 200 nM negative control siRNA; 72 h after transfection, total RNA isolated from these cells was subjected to qRT-PCR using a primer set specific to GNB4. d, Seventy-two hours after transfection, whole cellular lysate prepared from 182R-6 and TAMR-1 cells was transfected with either 200 nM DNMT3B siRNA or 200 nM negative control siRNA, and was subjected to Western blot analysis using antibodies against DNMT1, DNMT3A, DNMT3B and GNB4. Asterisk indicates p < 0.03
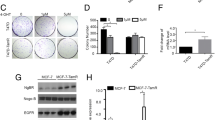
Ectopic expression of GNB4 enhanced proliferation of antiestrogen-resistant breast cancer cells in the presence of antiestrogen drugs
Since GNB4 was downregulated in antiestrogen-resistant breast cancer cells, we hypothesized that GNB4 may function as an “antidrug-resistant” gene that may restore the sensitivity of resistant cell lines to either fulvestrant or tamoxifen. To test our hypothesis by determining the role of GNB4 in the development of acquired fulvestrant and tamoxifen resistance in breast cancer, we generated GNB4 stable-expressing 182R-6 and TAMR-1 cell lines (Fig. 3a). Surprisingly, the MTT assay showed that the ectopic expression of GNB4 further enhanced drug-resistant features of these cells by significantly promoting their proliferation (p < 0.05, Fig. 3b and c), even though they were under treatment with either fulvestrant or tamoxifen. Interestingly, in the fulvestrant- and tamoxifen-free medium growth conditions, GNB4 overexpression had no effect on 182R-6 cell growth (Additional file 1: Figure S1A), but suppressed TAMR-1 cell proliferation (Additional file 1: Figure S1B). Our results suggest that GNB4 is a drug-resistant gene in fulvestrant- and tamoxifen-resistant breast cancer cells.

The ectopic expression of GNB4 enhances the proliferation of antiestrogen-resistant breast cancer cells. a, 182R-6 and TAMR-1 cells were transfected with either pEGFP-GNB4 or pEGFP-C1; after G418 selection, whole cellular lysate prepared from the positive cells was subjected to Western blot analysis using an antibody against GNB4. b and c, MTT assay was performed using 182R-6 (b) and TAMR-1 (c) cells stably expressing GNB4 or GFP as described in “Methods”. Asterisk indicates p < 0.05
Ectopic expression of GNB4 altered cell cycle and apoptosis of antiestrogen-resistant breast cancer cells
We next looked at the potential role of GNB4 in controlling the cell cycle and apoptosis of antiestrogen-resistant breast cancer cells. The cell-cycle analysis indicated that the ectopic expression of GNB4 shortened S-phase in 182R-6 cells and G2 in TAMR-1 cells (Fig. 4a and b). The apoptosis analysis showed that the enforced expression of GNB4 induced apoptosis of the 182R-6 cells, whereas it completely attenuated the induction of apoptosis in the TAMR-1 cells (Fig. 4c and d).

The ectopic expression of GNB4 causes alterations in cell cycle and apoptosis. a and b, 182R-6 and TAMR-1 cells stably expressing GNB4 or GFP grown to 90% confluency were subjected to cell cycle analysis using a GFP-Certified Nuclear-ID Red Cell Cycle Analysis Kit according to the manufacturer’s instructions. c and d, 182R-6 and TAMR-1 cells stably expressing GNB4 or GFP grown to 90% confluency were subjected to apoptosis analysis using an Annexin V-Cy3 Apoptosis Kit Plus according to the manufacturer’s instructions. Asterisk indicates p < 0.05
To understand the mechanism underlying the GNB4-mediated alterations in proliferation, cell cycle, and apoptosis, we determined the expression of cell cycle and apoptosis regulators in 182R-6 and TAMR-1 cells in response to a GNB4 expression. The Western blot analysis showed that cyclin A2 was downregulated, while CDK6 was upregulated in 182R-6 and TAMR-1 cells in response to the ectopic expression of GNB4 (Fig. 5a). Interestingly, GNB4 caused an induction in genes, including those of cyclin D1 and E, CDK2, BAX, and phosphorylated Akt1/2/3 in 182R-6 cells, whereas it attenuated the expression of these gene in TAMR-1 cells (Fig. 5a and b). Although BCL2 and phosphorylated ERK1/2 were elevated in 182R-6 by GNB4, they had no effect in TAMR-1 cells (Fig. 5b). Additionally, GNB4 had no effect on p21 expression in both cell lines (Fig. 5a).

Changes of cell cycle and apoptosis regulators in response to enforced GNB4 expression. a and b, Whole cellular lysate prepared from 182R-6 and TAMR-1 cells stably expressing GNB4 or GFP was subjected to Western blot analysis using the indicated antibodies
Knockdown of GNB4 with siRNA suppressed proliferation and induced apoptosis and cell cycle arrest of antiestrogen-resistant breast cancer cells
To further confirm the role of GNB4 in the control of cell proliferation, cell cycle and apoptosis in antiestrogen resistant 182R-6 and TAMR-1 cells, we then knocked down GNB4 using siRNA and measured the effect on cell proliferation, cell cycle and apoptosis. Western blot analysis showed that 72 h after transfection, the expression of GNB4 was attenuated in TAMR-1 cell line (Additional file 2: Figure S2), while had no effect in 182R-6 cell line. However, at 96 h after transfection, the expression of GNB4 was also reduced in 182R-6 cell line by GNB4 siRNA (Additional file 3: Figure S4). As expected, knockdown of GNB4 suppressed proliferation of 182R-6 and TAMR-1 cells in response to either fulvestrant or tamoxifen (Fig. 6a and b). Interestingly, Knockdown of GNB4 induced significantly apoptosis in TAMR-1 cells (Fig. 6b, middle panel), while had no effect on that in 182R-6 cells (Fig. 6a, middle panel). Furthermore, knockdown of GNB4 induced S-phase arrest in 182R-6 cells (Fig. 6a, right panel), whereas had no effect on that in TAMR-1 cells (Fig. 6b, right panel). Moreover, knockdown of GNB4 also inhibited proliferation of the parental S05 cells in the absence of antiestrogen drugs (Additional file 4: Figure S3), in addition to a role in drug resistance, may also implicating a role in the development of breast cancer.

Knockdown of GNB4 inhibits proliferation and induces cell cycle arrest and apoptosis. a and b, 182R-6 and TAMR-1 cells grown to 80% confluency were transiently transfected with either 30 nM GNB4 siRNA or 30 nM negative control siRNA; 24 h after transfection, the cells were replated in 96-well plate; the MTT assay was performed as described in “Methods”; 72 h (TAMR-1) or 96 h (182R-6) after transfection, the cells were harvested for cell cycle and apoptosis analyses. Asterisk indicates p < 0.05
Discussion
Although the well-established and effective endocrine therapy has provided millions of women with ER+ breast cancer with targeted treatment option [20, 23], most patients with metastatic disease would unfortunately and inevitably develop resistance to the drugs [28, 29], which has become a major clinical challenge in the treatment of this disease. As demonstrated, the reactivation (reestablishment) of ER and growth-factor signaling through crosstalk has been proposed as a primary mechanism for the development of antiestrogen resistance. A central role of upregulated EGFR/ErbB and the sustained activation of the EGFR/ErbB/ERK signaling pathway has been strongly indicated in the maintenance of antiestrogen-resistant breast cancer cell growth [30,31,32]. Although ERα is downregulated in tamoxifen-resistant breast cancer cell lines, the receptor is highly activated (phosphorylated) [32]. Importantly, the activated ER has been shown to promote the expression of insulin-like growth factor II (IGF2) in tamoxifen-resistant cell lines, resulting in the reactivation of insulin-like growth factor 1 receptor (IGF1R) signaling that activates EGFR via c-src-dependent phosphorylation [33]. Interestingly, the activated IGF2/IGF1R signaling may therefore in turn contribute to the phosphorylation of ER in tamoxifen-resistant cell lines [33], hence forming a positive-feedback proliferation loop. In addition, changes in the sensitivity of antiestrogen-resistant breast cancer cells to estradiol (E2) may also play a pivotal role in the development of antiestrogen resistance. Several lines of evidence have demonstrated that long-term estrogen deprivation (LTED) enhances the sensitivity of breast cancer cells to low levels of E2 (~ 10,000-fold reduction) [34,35,36]. The biological effect induced by E2 hypersensitivity, such as proliferation, was also mediated by the activation (phosphorylation) of ER and ERK1/2 (extracellular signal-regulated kinase 1/2) signalings. More recently, several studies highlighted a key role of protein kinases in the development of antiestrogen resistance, such as tyrosine kinase FYN and aurora kinase A [37, 38]. Interestingly, the tamoxifen-resistant breast cancer cells may derive from cancer stem-like cells [39].
To our knowledge, this study has revealed, for the first time, that GNB4 was epigenetically silenced in antiestrogen-resistant breast cancer cells, and it has highlighted an important role of GNB4 in the growth of antiestrogen resistant breast cancer cells. Heterotrimeric G proteins which are composed of an alpha subunit, a beta subunit (e.g. GNB4), and a gamma subunit function as molecular switches that play a crucial role in signal transduction from a cell surface receptor to internal effectors in a G protein-coupled receptor (GPCR) pathway [40]. Although the GPCR signaling pathway has been extensively studied, very little is known about GNB4. However, since the GPCR pathway plays a pivotal role in many biologic and pathologic processes, including tumorigenesis, it may also reflect a role of GNB4 in these processes. Evidence has demonstrated that GNB-isoforms are essential for chemokine-induced GPCR downstream signaling [41]. GNB4 may be one of the key genes that cause Charcot-Marie-Tooth disease, a heterogeneous group of the inherited neuropathies [42, 43]. GNB4 has also been linked to cancer. Haplotypes of GNB4 intron-1 have been shown to be associated with the survival rate of patients with colorectal and urothelial bladder carcinomas [44, 45]. We showed here that GNB4 was downregulated in both fulvestrant- and tamoxifen-resistant breast cancer cell lines, and that this was attributed to DNMT3B-mediated DNA methylation, because knockdown of DNMT3B by siRNA significantly elevated the expression of GNB4 at both mRNA and protein levels (Fig. 2). GNB4 has been reported to be downregulated in progressive breast cancer due to the acquired tamoxifen resistance [46], which is consistent with our results. Importantly, we found that the ectopic expression of GNB4 remarkably promoted the proliferation of antiestrogen-resistant breast cancer cells in response to antiestrogen drugs (Fig. 3). We also noted that the enforced expression of GNB4 caused cell cycles G2 and S to undergo phase acceleration (Fig. 4a and b), which may contribute to the GNB4-mediated proliferation of antiestrogen-resistant cells (Fig. 3b and c). Interestingly, the ectopic expression of GNB4 completely abolished apoptosis in tamoxifen-resistant TAMR-1 cells, while it significantly induced apoptosis in fulvestrant-resistant 182R-6 cells (Fig. 4c and d). However, the dramatic effects of GNB4 on apoptosis in both cell lines may all contribute to GNB4-mediated cellular proliferation (Fig. 3b and c). Recently, several interesting studies have indicated that apoptotic cells could promote the proliferation of surrounding cells (apoptosis-induced proliferation) due to the mitogenic signals released by the apoptotic cells [47,48,49]. siRNA-mediated GNB4 knockdown, however, suppressed proliferation of 182R-6 and TAMR-1 cells in the presence of antiestrogen drugs, and induced S-phase arrest of 182R-6 cells and apoptosis of TAMR-1 cells (Fig. 6a and b), further validating a crucial role of GNB4 in the development of antiestrogen resistance of breast cancer cells. GNB4 siRNA had no effect on GNB4 expression in 182R-6 cells at 72 h after transfection (Additional file 2: Figure S2), may reflect a longer half-life time of GNB4 in this cell line, since in another independent experiment, GNB4 was noted to be downregulated at 96 h after transfection (Additional file 3: Figure S4A and B).
GNB4 is a key component of heterotrimeric G proteins, which play an essential role in the transduction of GPCR-mediated signaling. Because of the crucial role of GPCR in the activation of AKT (v-AKT murine thymoma viral oncogene homolog) and ERK1/2 pathways [50, 51], an increase in phosphorylated AKT and/or phosphorylated ERK1/2 was expected to be seen in antiestrogen-resistant cells in response to the ectopic GNB4 expression. As expected, the phosphorylated AKT and phosphorylated ERK1/2 were elevated in 182R-6 cells in response to the GNB4 expression (Fig. 5b). This may contribute to GNB4-mediated cellular proliferation (Fig. 3b). However, the enforced expression of GNB4 caused a reduction in the phosphorylated AKT and had no effect on the phosphorylated ERK1/2 in TAMR-1 cells (Fig. 5b). The mechanism involved is unclear. It may be interesting to look at the crosstalk with other pathways, such as ER. Importantly, the GNB4-induced upregulation of BAX in 182R-6 cells and the downregulation in TAMR-1 cells may contribute to GNB4’s effect on apoptosis in these cells (Fig. 5b and Fig. 4c and d). We also noted that GNB4 caused an induction in BCL2 in 182R-6 cells, while it had no effect in TAMR-1 cells (Fig. 5b), implicating that the functional effect of GNB4 on apoptosis in 182R-6 cells may be due to the balance between proapoptotic (BAX) and antiapoptotic (BCL2) proteins.
Cyclins and cyclin-dependent kinases control cell-cycle progression and transitions. Cyclin A interacts with cyclin-dependent kinase 2 (CDK2) or CDK1 to form a complex that governs the S phase of the cell cycle [52]. Cyclin D interacts with CDK4 or CDK6 to form a complex that controls the G1 phase of the cell cycle [49]. However, in a complex with CDK2, cyclin E controls the S-phase progression and G1-S transition [52, 53]. The results we presented here showed that cyclin E and CDK2 were upregulated in 182R-6 cells in response to GNB4 (Fig. 5a), which may contribute to the shortened S phase (Fig. 4a). The ectopic GNB4, however, attenuated the expression of CDK2 and cyclin A and E in TAMR-1 cells (Fig. 5a), which may contribute to the S-phase arrest (Fig. 4a). Although the ectopic GNB4 caused a profound induction in both cyclin D1 and CDK6 in 182R-6 cells (Fig. 5a), this induction had no effect on the G1 phase of the cell cycle (Fig. 4a). The GNB4-induced upregulation of CDK6 and downregulation of cyclin D1 had no effect on the G1 phase of TAMR-1 cells (Fig. 5a and Fig. 4b). We also noted that the ectopic GNB4 had no effect on the expression of p21, an inhibitor of cyclin D/CDK6 and cyclin E/CDK2 complexes [54]. However, the mechanism underlying the shortened GNB4-induced G2 phase of the cell cycle in TAMR-1 cells is still unclear. Although we did not measure the expression of CDC25 and speedy/ringo C in these cells, it has been demonstrated that these two molecules play an important role in controlling G2-phase progression [55, 56].
Conclusion
In summary, although the observed reduced level of GNB4 in the two antiestrogen resistant cell lines is not the underlying cause of antiestrogen resistance, GNB4 is important for growth of both antiestrogen resistant and antiestrogen sensitive breast cancer cells and thereby a target for treatment of breast cancer.
Abbreviations
- AKT1:
-
V-AKT murine thymoma viral oncogene homolog 1
- BAX:
-
BCL2-associated X protein
- BCL2:
-
B-cell CLL/Lymphoma 2
- CDK2:
-
cyclin-dependent kinase 2
- CDK6:
-
cyclin-dependent kinase 6
- DBD:
-
DNA-binding domain
- DNMT1:
-
DNA methyltransferase 1
- DNMT3A:
-
DNA methyltransferase 3A
- DNMT3B:
-
DNA methyltransferase 3B
- ER:
-
Estrogen receptor
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase gene
- GNB4:
-
Guanine nucleotide-binding protein beta-4
- HMEC:
-
Human mammary epithelial cells
- LBD:
-
Ligand-binding domain
- MeCP2:
-
Methyl-CpG-binding protein 2
- MTT:
-
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NTD:
-
N-terminal domain
- qRT-PCR:
-
Quantitative real-time RT-PCR
- siRNA:
-
Small interfering RNA
References
Schairer C, Mink PJ, Carroll L, Devesa SS. Probabilities of death from breast cancer and other causes among female breast cancer patients. J Natl Cancer Inst. 2004;96(17):1311–21.
Russo IH, Russo J. Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia. 1998;3(1):49–61.
Sommer S, Fuqua SA. Estrogen receptor and breast cancer. Semin Cancer Biol. 2001;11(5):339–52.
Keshamouni VG, Mattingly RR, Reddy KB. Mechanism of 17-beta-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER2 AND PKC-delta. J Biol Chem. 2002;277(25):22558–65.
Kovalchuk O, Tryndyak VP, Montgomery B, Boyko A, Kutanzi K, Zemp F, et al. Estrogen-induced rat breast carcinogenesis is characterized by alterations in DNA methylation, histone modifications and aberrant microRNA expression. Cell Cycle. 2007;6(16):2010–8.
Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer. 2014;14(1):26–38.
Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–86.
Dey P, Barros RP, Warner M, Strom A, Gustafsson JA. Insight into the mechanisms of action of estrogen receptor beta in the breast, prostate, colon, and CNS. J Mol Endocrinol. 2013;51(3):T61–74.
Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol. 1999;13(10):1672–85.
Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122(1):33–43.
Zhao C, Gao H, Liu Y, Papoutsi Z, Jaffrey S, Gustafsson JA, et al. Genome-wide mapping of estrogen receptor-beta-binding regions reveals extensive cross-talk with transcription factor activator protein-1. Cancer Res. 2010;70(12):5174–83.
Maggiolini M, Bonofiglio D, Marsico S, Panno ML, Cenni B, Picard D, et al. Estrogen receptor alpha mediates the proliferative but not the cytotoxic dose-dependent effects of two major phytoestrogens on human breast cancer cells. Mol Pharmacol. 2001;60(3):595–602.
Sun PM, Gao M, Wei LH, Mustea A, Wang JL, Konsgen D, et al. An estrogen receptor alpha-dependent regulation of estrogen receptor-related receptor alpha in the proliferation of endometrial carcinoma cells. Int J Gynecol Cancer. 2006;16(Suppl 2):564–8.
Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;64(1):423–8.
Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A. 2004;101(6):1566–71.
Stanford JL, Szklo M, Brinton LA. Estrogen receptors and breast cancer. Epidemiol Rev. 1986;8:42–59.
Hayes EL, Lewis-Wambi JS. Mechanisms of endocrine resistance in breast cancer: an overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015;17:40.
Moy B, Goss PE. Estrogen receptor pathway: resistance to endocrine therapy and new therapeutic approaches. Clin Cancer Res. 2006;12(16):4790–3.
Luzhna L, Lykkesfeldt AE, Kovalchuk O. Altered radiation responses of breast cancer cells resistant to hormonal therapy. Oncotarget. 2015;6(3):1678–94.
Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805–16.
Nicholson RI, Johnston SR. Endocrine therapy--current benefits and limitations. Breast Cancer Res Treat. 2005;93(Suppl 1):S3–10.
Johnston SR, Martin LA, Dowsett M. Life following aromatase inhibitors--where now for endocrine sequencing? Breast Cancer Res Treat. 2005;93(Suppl 1):S19–25.
Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a north American trial. J Clin Oncol. 2002;20(16):3386–95.
Lykkesfeldt AE, Briand P. Indirect mechanism of oestradiol stimulation of cell proliferation of human breast cancer cell lines. Br J Cancer. 1986;53(1):29–35.
Lykkesfeldt AE, Madsen MW, Briand P. Altered expression of estrogen-regulated genes in a tamoxifen-resistant and ICI 164,384 and ICI 182,780 sensitive human breast cancer cell line, MCF-7/TAMR-1. Cancer Res. 1994;54(6):1587–95.
Wang B, Chen J, Santiago FS, Janes M, Kavurma MM, Chong BH, et al. Phosphorylation and acetylation of histone H3 and autoregulation by early growth response 1 mediate interleukin 1beta induction of early growth response 1 transcription. Arterioscler Thromb Vasc Biol. 2010;30(3):536–45.
Wang B, Li D, Sidler C, Rodriguez-Juarez R, Singh N, Heyns M, et al. A suppressive role of ionizing radiation-responsive miR-29c in the development of liver carcinoma via targeting WIP1. Oncotarget. 2015;6(12):9937–50.
Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–43.
Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11(4):643–58.
Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68(3):826–33.
Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009;114(2):263–75.
Thrane S, Lykkesfeldt AE, Larsen MS, Sorensen BS, Yde CW. Estrogen receptor alpha is the major driving factor for growth in tamoxifen-resistant breast cancer and supported by HER/ERK signaling. Breast Cancer Res Treat. 2013;139(1):71–80.
Nicholson RI, Hutcheson IR, Britton D, Knowlden JM, Jones HE, Harper ME, et al. Growth factor signalling networks in breast cancer and resistance to endocrine agents: new therapeutic strategies. J Steroid Biochem Mol Biol. 2005;93(2–5):257–62.
Masamura S, Santner SJ, Heitjan DF, Santen RJ. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995;80(10):2918–25.
Martin LA, Farmer I, Johnston SR, Ali S, Dowsett M. Elevated ERK1/ERK2/estrogen receptor cross-talk enhances estrogen-mediated signaling during long-term estrogen deprivation. Endocr Relat Cancer. 2005;12(Suppl 1):S75–84.
Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278(33):30458–68.
Elias D, Vever H, Lænkholm AV, Gjerstorff MF, Yde CW, Lykkesfeldt AE, et al. Gene expression profiling identifies FYN as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene. 2015;34(15):1919–27.
Thrane S, Pedersen AM, Thomsen MB, Kirkegaard T, Rasmussen BB, Duun-Henriksen AK, et al. A kinase inhibitor screen identifies Mcl-1 and aurora kinase a as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. 2015;34(32):4199–5210.
Lin X, Li J, Yin G, Zhao Q, Elias D, Lykkesfeldt AE, et al. Integrative analyses of gene expression and DNA methylation profiles in breast cancer cell line models of tamoxifen-resistance indicate a potential role of cells with stem-like properties. Breast Cancer Res. 2013;15(6):R119.
Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9(1):60–71.
Block H, Stadtmann A, Riad D, Rossaint J, Sohlbach C, Germena G, et al. Gnb isoforms orchestrate a signaling pathway comprising Rac1 and Plcbeta2/3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood. 2016;127(3):314–24.
Soong BW, Huang YH, Tsai PC, Huang CC, Pan HC, Lu YC, et al. Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate Charcot-Marie-tooth disease. Am J Hum Genet. 2013;92(3):422–30.
Baets J, De Jonghe P, Timmerman V. Recent advances in Charcot-Marie-tooth disease. Curr Opin Neurol. 2014;27(5):532–40.
Riemann K, Struwe H, Alakus H, Obermaier B, Schmitz KJ, Schmid KW, et al. Association of GNB4 intron-1 haplotypes with survival in patients with UICC stage III and IV colorectal carcinoma. Anticancer Res. 2009;29(4):1271–4.
Riemann K, Struwe H, Eisenhardt A, Obermaier B, Schmid KW, Siffert W. Characterization of intron-1 haplotypes of the G protein beta 4 subunit gene--association with survival and progression in patients with urothelial bladder carcinoma. Pharmacogenet Genomics. 2008;18(11):999–1008.
Umar A, Kang H, Timmermans AM, Look MP, Meijer-van Gelder ME, et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol Cell Proteomics. 2009;8(6):1278–94.
Ryoo HD, Bergmann A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol. 2012;4(8):a008797.
Dichtel-Danjoy ML, Ma D, Dourlen P, Chatelain G, Napoletano F, Robin M, et al. Drosophila p53 isoforms differentially regulate apoptosis and apoptosis-induced proliferation. Cell Death Differ. 2013;20(1):108–16.
Mollereau B, Perez-Garijo A, Bergmann A, Miura M, Gerlitz O, Ryoo HD, et al. Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ. 2013;20(1):181.
Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem. 1998;273(30):19080–5.
Wang B, Hendricks DT, Wamunyokoli F, Parker MI. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006;66(6):3071–7.
Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–93.
Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell. 1994;77(1):107–20.
Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signalling. EMBO J. 2012;31(12):2705–13.
Ogura Y, Sakaue-Sawano A, Nakagawa M, Satoh N, Miyawaki A, Sasakura Y. Coordination of mitosis and morphogenesis: role of a prolonged G2 phase during chordate neurulation. Development. 2011;138(3):577–87.
Cheng A, Solomon MJ. Speedy/Ringo C regulates S and G2 phase progression in human cells. Cell Cycle. 2008;7(19):3037–47.
Acknowledgements
We thank Ms. Rommy Rodriguez-Juarez, and Dr. Andrey Golubov and Dr. Yaroslav Ilnytskyy for their technical and bioinformatics support. Ms. Megan Malach was a recipient of the AI-HS Summer Studentship. Ms. Emily Carpenter was a recipient of the NSERC Summer Research Award.
Funding
Study was supported by the Alberta Cancer Foundation and the CIHR grants to Dr. Olga Kovalchuk. The funding body did not have any influence on the design of the study, data collection, analysis or interpretation or writing the manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due to the ongoing IP-protection issues but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
BW, JF, AEL and OK conceived and designed the study; BW, DL, RRJ, AF, QS, MM, EC and JF performed the experiments and collected data; BW, DL and JF analyzed and interpreted data; BW, JF, AEL and OK drafted and revised the manuscript; BW and OK supervised the study; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consented for publication of the manuscript in the journal of BMC Cancer.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Effect of GNB4 overexpression on cell growth of 182R-6 and TAMR-1 cell lines (no drug treatment). A and B, MTT assay was performed using 182R-6 (a) and TAMR-1 (b) cells stably expressing GFP or GNB4 as described in “Methods”, using fulvestrant- and tamoxifen-free medium. Asterisk indicates p < 0.05. (PPTX 42 kb)
Additional file 2:
Figure S2. Knockdown of GNB4 in TAMR-1 and 182R-6 cells using siRNA. 182R-6 and TAMR-1 cells grown to 80% confluency were transiently transfected with either 30 nM GNB4 siRNA or 30 nM negative control siRNA; at 72 h after transfection, whole cellular lysates were prepared and subjected to Western blot analysis using antibody against GNB4. (PPTX 508 kb)
Additional file 3:
Figure S4. siRNA-mediated knockdown of GNB4 in TAMR-1 and 182R-6 cells. A, 182R-6 and TAMR-1 cells grown to 80% confluency were transiently transfected with either 30 nM GNB4 siRNA or 40 nM negative control siRNA; At 72 and 96 h after transfection, whole cellular lysates were prepared and subjected to Western blot analysis using antibody against GNB4. B, a relative densitometry (GNB4/Actin) was performed to further validate the GNB4 expression in 182R-6 cell line 96 h after transfection, using ImageJ software. Asterisk indicates p < 0.05. (PPTX 49 kb)
Additional file 4:
Figure S3. Knockdown of GNB4 using siRNA suppresses proliferation of parental S05 cells. S05 cells grown to 80% confluency were transiently transfected with either 30 nM GNB4 siRNA or 30 nM negative control siRNA; at 24 h after transfection, the cells were replated in 96-well plate, MTT assay was performed as described in “Methods”. Asterisk indicates p < 0.05. (PPTX 37 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, B., Li, D., Rodriguez-Juarez, R. et al. A suppressive role of guanine nucleotide-binding protein subunit beta-4 inhibited by DNA methylation in the growth of anti-estrogen resistant breast cancer cells. BMC Cancer 18, 817 (2018). https://doi.org/10.1186/s12885-018-4711-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4711-0