
Abstract
Background
The higher specificity of amino-acid positron emission tomography (AA-PET) in the diagnosis of gliomas, as well as in the differentiation between recurrence and treatment-related alterations, in comparison to contrast enhancement in T1-weighted MRI was demonstrated in many studies and is the rationale for their implementation into radiation oncology treatment planning. Several clinical trials have demonstrated the significant differences between AA-PET and standard MRI concerning the definition of the gross tumor volume (GTV). A small single-center non-randomized prospective study in patients with recurrent high grade gliomas treated with stereotactic fractionated radiotherapy (SFRT) showed a significant improvement in survival when AA-PET was integrated in target volume delineation, in comparison to patients treated based on CT/MRI alone.
Methods
This protocol describes a prospective, open label, randomized, multi-center phase II trial designed to test if radiotherapy target volume delineation based on FET-PET leads to improvement in progression free survival (PFS) in patients with recurrent glioblastoma (GBM) treated with re-irradiation, compared to target volume delineation based on T1Gd-MRI. The target sample size is 200 randomized patients with a 1:1 allocation ratio to both arms. The primary endpoint (PFS) is determined by serial MRI scans, supplemented by AA-PET-scans and/or biopsy/surgery if suspicious of progression. Secondary endpoints include overall survival (OS), locally controlled survival (time to local progression or death), volumetric assessment of GTV delineated by either method, topography of progression in relation to MRI- or PET-derived target volumes, rate of long term survivors (>1 year), localization of necrosis after re-irradiation, quality of life (QoL) assessed by the EORTC QLQ-C15 PAL questionnaire, evaluation of safety of FET-application in AA-PET imaging and toxicity of re-irradiation.
Discussion
This is a protocol of a randomized phase II trial designed to test a new strategy of radiotherapy target volume delineation for improving the outcome of patients with recurrent GBM. Moreover, the trial will help to develop a standardized methodology for the integration of AA-PET and other imaging biomarkers in radiation treatment planning.
Trial registration
The GLIAA trial is registered with ClinicalTrials.gov (NCT01252459, registration date 02.12.2010), German Clinical Trials Registry (DRKS00000634, registration date 10.10.2014), and European Clinical Trials Database (EudraCT-No. 2012-001121-27, registration date 27.02.2012).
Similar content being viewed by others


Background
During the last years enormous progress has been made in the area of high precision radiotherapy [1]. In the brain it is now technically feasible to irradiate complex target volumes with a precision of less than 1 mm, while sparing normal tissues [2]. This offers the opportunity to significantly escalate the radiation dose for the tumor tissue, which is considered to be a key for increasing local control rates. However, the potential of high precision radiotherapy can only be realized when the tumor volume can be accurately delineated by imaging techniques [3]. Studies have shown that standard anatomic imaging modalities (CT, MRI), while very accurate at visualizing normal anatomical structures, are limited in defining tumor extension for radiation treatment planning [4]. Traditionally, the target volume definition for irradiation, as well as re-irradiation after recurrence, of malignant gliomas is based on T1-weighted MRI with Gadolinium (Gd) [5]. Contrast enhancement is a consequence of disruption of the blood-brain barrier (BBB) which does not necessarily reflect the real tumor extension in gliomas. Gross tumor mass has been detected beyond the margins of contrast enhancement, in the surrounding edema and even in the adjacent normal-appearing brain tissue [6–10].
After therapy (surgery, irradiation and/or chemotherapy), BBB disturbances can frequently be treatment-related (for example associated with postoperative granulation or radiation necrosis) and cannot be differentiated from persistent tumor on conventional MRI [9]. This phenomenom was termed “pseudoprogression” [11] and, in this case non-tumoral tissue may be erroneously included in the gross tumor volume (GTV), leading to a higher rate of sides effects after re-irradiation. Vice versa, after systemic treatment with vascular endothelial growth factor (VEGF) receptor signalling pathway inhibitors such as bevacizumab, “pseudoresponse” has been described [11–13].
In patients that have been previously irradiated, the volume of normal tissue included in high dose areas should be as small as possible [14] to avoid severe toxicities, such as radiation necrosis [15]. Therefore, the target volume has to encompass mainly the macroscopic tumor mass (GTV) without including a large area of suspected microscopic tumor infiltration (clinical target volume, CTV). The margins of the planning target volume (PTV) have to be very small, in order to spare normal brain tissue. High conformal radiation strategies like stereotactic-fractionated radiotherapy (SFRT), image-guided radiotherapy (IGRT) and intensity modulated radiotherapy (IMRT) are used to focus the irradiation on the gross tumor mass and reduce the required margin, making GTV delineation in this case a major issue [9, 16, 17].
For high precision radiotherapy, inaccuracies in tumor delineation may offset any gain in local control rates achieved by dose escalation, emphasizing the need for new imaging approaches to increase tumor delineation for high precision radiotherapy [18].
Along this line, imaging the biological and molecular characteristics of the tumor tissue by positron emission tomography (PET) is an interesting approach to improve treatment planning for high precision radiotherapy. Multiple studies correlating imaging findings with histopathological evaluation in surgically treated patients with high grade glioma have indicated that molecular imaging with amino acid (AA) PET (L-[methyl-11C]methionine (MET) or O-(2-[18F]fluoroethyl)-L-tyrosine (FET)) is more specific and equally sensitive for tumor staging than MRI [4, 19]. Based on these data, the infrastructure for AA-PET imaging has become widely available in major hospitals [20]. Although AA-PET imaging shows great promise for target delineation, it has not been rigorously evaluated in clinical trials. Several studies in patients with gliomas have indicated that PET based target volumes differ markedly from target volumes defined by MRI, but the methodology for tumor delineation on PET images differs significantly among these studies [8, 21–28]. More importantly, there are no randomized trials that have evaluated the impact of PET based radiotherapy on patient outcome. According to relevant clinical trial registers, no clinical trials are currently running or planned for this indication (http://www.drks.de, http://www.controlled-trials.com, http://clinicaltrials.gov, http://www.who.int/ictrp; last accessed on 07.05.2016). Generally, imaging techniques have so far not been evaluated with the same rigor as therapeutic agents. The high costs associated with modern imaging techniques make it necessary to use a similar approach as for evaluation of new therapeutic agents.
The hypothesis of the study is that AA-PET, having a higher specificity and equal sensitivity for tumor tissue in comparison to MRI (T1 with gadolinium), will visualize the tumor mass with a higher precision and thus will improve patient outcome. This hypothesis was pre-tested in a previous small prospective monocentric non-randomized pilot study led by the principal investigators [9]. In this cohort of 44 patients, a statistically significant better survival time was reported in patients with recurrent gliomas treated with re-irradiation using SFRT based on AA-PET in comparison to the same irradiation regime based on T1Gd-MRI on univariate analysis. The goal of this trial is to verify the improved outcome for patients in a randomized multicenter phase II study with progression free survival (PFS) as the primary endpoint to specifically address the potential impact of the differences in radiation target volumes. The results of this trial could have a significant impact not only on GTV delineation in recurrent GBM, but also in the tumor mass delineation of primary tumors, in the evaluation of tumor response and treatment monitoring and in developing of a standardized methodology for target volume delineation based on PET. A further goal of the study is to establish a framework for the use of molecular imaging in radiation oncology.
Methods/design
Trial design and setting
This is a prospective, open label randomized (allocation 1:1) two-arm parallel group phase II multi-centre trial designed to test for differences in the impact of an FET-PET-based (experimental, Arm A) versus a T1Gd-MRI-based (control, Arm B) treatment planning on the progression-free survival in patients with recurrent GBM treated with re-irradiation.
Trial sites are academic hospitals and community clinics located in Aachen, Bonn, Dessau, Erlangen, Freiburg, Hannover (two sites), Karlsruhe, Köln, Magdeburg, Mannheim, Marburg, München, Offenburg, Rostock, Stuttgart, Trier, and Tübingen.
The trial was approved by the ethics committee of the University of Freiburg (EK-Freiburg 133/10) and by the local ethics committees of participating sites. The GLIAA trial has been thoroughly examined and approved by the Federal Office for Radiation Protection (Bundesamt für Strahlenschutz, BfS) and the Federal Institute for Drugs and Medical Devices (Bundesamt für Arzneimittel und Medizinprodukte, BfArM). Written informed consent for study participation is obtained from all patients before the initiation of any study-specific procedures. The GLIAA trial is associated with the German Neurooncological Network (Neuroonkologische Arbeitsgemeinschaft, NOA-10) and Working Group Radiation Oncology (Arbeitsgemeinschaft Radiologische Onkologie, ARO2013-1) of the German Cancer Society (Deutsche Krebsgesellschaft, DKG).
Study population
The target population for this trial is previously irradiated patients with recurrent GBM. For the proposed trial, there is no gender requirement. No gender ratio has been stipulated in this study as the results of the preclinical and/or clinical studies did not indicate any difference in the effect of the study treatment in terms of efficacy and safety. No healthy persons will be included. To avoid selection bias, investigators should enroll patients irrespectively of whether or not any differences are seen in GTV-delineation based on FET-PET versus T1Gd-MRI.
Key inclusion criteria at the time of randomization are: (1) Patient’s written informed consent (IC) obtained latest the day after FET-PET acquisition, (2) legal capacity: Patient is able to understand the nature, significance and consequences of the study, (3) age ≥ 18 years (no upper limit of age), (4) Karnofsky Performance Score (KPS) > 60 %, (5) registration in the electronic case report form, (6) recurrence of GBM (WHO grade IV) and either not eligible for tumor resection or with macroscopic residual tumor after resection of the recurrent GBM, (7) histological confirmation of GBM at initial or secondary diagnosis, (8) previous radiation therapy of high grade glioma (WHO Grad III or IV) with a total dose of 59.4 - 60Gy (single dose 1.8 – 2.0 Gy), (9) at least 6 months between the end of pre-irradiation and randomization, (10) recurrent tumor visible on FET-PET and T1Gd-MRI with the maximum diameter measuring 1 cm to 6 cm by either technique (in case of multifocal tumor, the sum of all diameters has to be 1-6 cm on FET-PET and T1Gd-MRI), (11) target volume definition possible according to both study arms, (12) start of re-irradiation planned within 2 weeks from FET-PET and MRI. Key Exclusion criteria are: (1) Recent (≤ 4 weeks before IC) histological result showing no tumor recurrence, (2) previous treatment of GBM with bevacizumab or other molecular targeted therapies less than 6 months before MRI and FET-PET used for radiotherapy planning, (3) technical impossibility to use MRI or FET-PET dataset for RT planning, (4) less than 2 weeks between the last day of last chemotherapy given and planned start of reirradiation, (5) less than 3 weeks between resection of recurrent GBM and planned start of re-irradiation, (6) chemotherapy or molecular targeted therapies planned during re-irradiation, (7) additional chemotherapy or molecular targeted therapy or further surgery planned before diagnosis of further tumor progression after study intervention, (8) simultaneous participation in other interventional trials which could interfere with this trial and/or participation in a clinical trial within the last thirty days before the start of this study and/or previous participation (randomization) in this study, (9) pregnancy, nursing, or patient not willing to prevent a pregnancy during treatment, (10) known or persistent abuse of medication, drugs or alcohol, (11) known allergy against the MRI contrast agent Gadolinium or the PET tracer 18F-FET or against any of the components.
Study treatment and procedures
The trial randomizes the patients to the following two treatment arms with a 1:1 allocation ratio: In the control intervention, the MRI based GTV delineation will be performed with respect only to the T1Gd-MRI image dataset. Here, the GTV is defined as the tumor related contrast enhancement with no safety margin in accordance with a neuroradiologist. In the experimental arm, the PET based GTV delineation will employ FET uptake with a greater value than 1.8 +/- 0.1 times of normal brain tissue uptake as a starting point, visually verified and modified by an experienced nuclear medicine specialist together with the treating radiation oncologist. To define the acquisition protocol and reconstruction parameters for each participating study centre, PET/CT scanner phantom studies will be performed. Freiburg will be the reference centre.
Starting from the GTV, for both target volumes (MRI and FET-PET based), a CTV will be defined by adding 3 mm in either direction respecting anatomical boundaries like skull and/or tentorium. This CTV will then be expanded to the PTV by adding 1–2 mm in all directions.
Radiotherapy planning will be performed after randomization using the predefined target volume for the treatment arm allocated. In both study arms, patients will be given a high-precision re-irradiation with a total dose 39 Gy, 3 Gy/d, 5×/week to the PTV as defined according to the respective study arm. The dose specification will be according to the criteria of the International Commission on Radiation Units and Measurements (ICRU) with the 95 % isodose surrounding the PTV.
In both treatment arms, the MRI/CT based contouring of risk organs will be done after the definition of the GTV. The following constraints for normal tissue/organs at risk have to be respected in radiotherapy planning, which relate to the cumulative doses from the actual and the prior radiation treatment in the same irradiated point / area, calculated by the equivalent dose in 2 Gy fractions (EQD2, α/β = 2 Gy): (1) The maximum total dose to the optical chiasm and/or both optical nerves must not exceed 54 Gy, (2) if only one optical nerve is involved, the maximum dose must not exceed 60 Gy, (3) the maximum dose to the brain stem must not exceed 66 Gy in 10 % of volume, (4) the medulla oblongata must not receive more than 60 Gy at maximum point, (5) the maximum dose to the retina must not exceed 54 Gy.
Irradiation is given either as SFRT using an external coordinate system and/or IGRT with CT and/or kV imaging of the treatment position before every treatment fraction. The mean positioning tolerance of all fractions as documented by this imaging must be ≤ 2 mm.
The flowchart of the treatment and follow-up schedule is shown in Fig. 1.

Flowchart of the GLIAA trial. AA-PET = amino acid positron emission tomography; MRI = magnetic resonance imaging; FET = O-(2-[18F]fluoroethyl)-L-tyrosine; Gd = gadolinium
Quality assurance for radiation treatment planning
After completion of radiotherapy planning for the first study patient, the study centers will upload the pseudonymized PET, CT and MRI images well as the pseudonymized radiotherapy treatment plan on a dedicated web-platform. A review committee including a radiation oncologist, a nuclear medicine physician and a physicist in the coordinating study centre in Freiburg will check the key parameters of imaging and treatment planning before the initiation of radiotherapy of the second study patient. After passing quality assurance for the at least one patient, further imaging and RT planning will undergo mutual monitoring by the members of the study group in the framework of regular study group meetings.
Study endpoints
The primary endpoint is PFS, defined as time from randomization until tumor progression or death. Progression is determined based on MRI and confirmed by AA-PET and/or positive biopsy/surgery as follows. A tumor-suspicious lesion on MRI according to RANO criteria [29] which is confirmed by AA-PET and/or biopsy/surgery is considered as tumor progression, while a negative PET-scan will exclude progression. A positive PET scan, if unclear, should be followed by biopsy. Patients receiving any new treatment (chemotherapy or immunotherapy) for progression of their GBM in the absence of diagnosed tumor progression, and patients receiving surgery for distant progression or receiving bevacizumab for a radionecrosis will also be considered as having an event for the endpoint PFS, at the date that treatment was initiated.
Secondary endpoints are the following. (1) Overall survival (OS) is defined as time from randomization to death. (2) Volumetric assessment of GTV based on PET and MRI is based on PET/MRI + RT-structure set image co-registration. The relation of both GTVs to each other, especially overlap and non-overlap volumes, will be assessed. (3) The topography of progression after re-irradiation will be evaluated at the time of progression. In all available image datasets, the topographical relation of the tumor re-growth will be scored as local progression or distant progression: Local progression will be determined as in field progression (largest proportion within the PTV), or margin progression (largest proportion within 2 cm and located in the same anatomical region as the PTV); Distant progression will be determined as outfield progression (largest proportion clearly [>2 cm] outside of PTV or located in another anatomical region). Progression will be judged as either in PET/MRI-GTV, marginal to PET/MRI-GTV, or clearly outside PET/MRI-GTV, both for PET and MRI, respectively. (4) Locally controlled survival is defined as time from randomization to local progression (see 3) or death. (5) Long term survival is defined as OS > 1 year. (6) The topography of necrosis after re-irradiation will be determined with respect to the irradiated volume (PTV) as described above. (6) Quality of life (QoL) will be determined using the EORTC QLQ-C15 PAL questionnaire [30] which is especially aimed to patients in a (near) palliative care setting. The primary scale for the QoL evaluation is the global health status/QoL scale status. The other scales and single items are secondary outcomes. The primary outcome measure is the change from baseline before radiation therapy to follow up measurements. (7) A possible impact of diffusion/perfusion and FLAIR MRI on target volume delineation will be analyzed optionally in departments having the technology and the know-how for these investigations. These evaluations will be handled as a separate subproject, to be described elsewhere. (8) Adverse events occurring until incl. day 7 after FET-PET imaging are registered to address safety of FET-PET imaging. (9) Occurrence of a range of pre-specified adverse events (AEs) considered as possible radiotherapy treatment toxicity is registered and documented according to NCI CTCAE v4.03 (http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf) during treatment and follow up. Events occurring more than 90 days after the start of irradiation are considered as late toxicity. In addition, we register serious adverse events (SAEs) occurring from start of radiotherapy until 30 days after end of radiotherapy as well as SAEs occurring during the complete follow up period which are considered as related to radiotherapy.
Sample size
The number of patients to recruit to this phase II study is derived from the primary endpoint PFS. Based on data from a literature report [31], where 6-months PFS ranged between 28 and 39 %, and considering that this study includes also patients with larger tumors up to a diameter of 6 cm, we expect 30 % PFS at 6 months in the control arm. The objective is to detect a difference if the experimental treatment entails an increase of at least 15 % in the 6-months PFS rate. Such an improvement seems feasible, since OS in the pilot study was 5 months in the control arm (treated based on MRI) versus 9.5 months in the experimental arm (treated based on PET/SPECT) [9]. Assuming exponentially distributed PFS times, the target difference of a 15 % increase to 45 % 6-months PFS corresponds to a hazard ratio (HR) of 0.667 for the experimental versus the control group (median ratio of 1.5).
The study was planned under these assumptions, using the comparative phase II design with one-sided type I error rate α = 10 and 90 % power proposed by Korn et al. [32], and a group sequential plan with one interim analysis during the recruitment phase, with the option to stop the trial early for futility. Assuming a constant recruitment rate, the following procedure to test the null hypothesis H0: HR ≥ 1.0 against the alternative hypothesis of superiority in the experimental arm, H1: HR < 1.0, has the desired properties (SAS Version 9.2, proc sequ design): After recruitment of 115 patients over a period of 15 months, the trial is to be stopped for futility (accept H0) if the p-value of the one-sided log-rank test of H0 versus H1 is above 0.51742. Otherwise, recruitment is to continue for another 9 months up to a total of 184 patients over a total period of 24 months, with additional follow up for 12 months. At the final analysis after 36 months, a log-rank test of H0 versus H1 at one-sided nominal level of α = 10 % is performed. With this sequential plan, the entire procedure, accounting for the interim analysis, has a power of 90 % and a type I error rate of 9.252 %. If H1 is true, the probability to stop for futility is 3.021 %. Under H1, the expected number of events is 71.85 at 15 months and 176.22 at 36 months. To compensate for possible losses to follow up or ineligible patients, a target number of 200 randomizations over a period of 24 months was planned, and the interim analysis should be performed when 125 = 200 × 15/24) patients would be randomized.
Randomization
Central randomization is performed by means of the minimization technique with a random element as initially described by Pocock and Simon [33], using a computerized randomizer tool (https://www.randomizer.at/) with the following factors in the minimization algorithm: (1) time since first radiation treatment, calculated between the last day of previous irradiation and randomization (≤14 months vs. >14 months) (2) previous chemotherapy treatment (≤ 7 cycles of TMZ vs. > 7 cycles) (3) maximum tumor diameter on MRI (GTV ≤ 3 cm vs. GTV > 3 cm) (4) MGMT-status (methylated vs. non-methylated vs. not yet determined). An open label design was chosen because effective blinding was considered unfeasible. However, the randomization procedure guarantees concealment of treatment allocation und thus minimizes selection bias.
Statistical analysis
Because this is a phase II study, the primary analyses of the efficacy endpoints PFS, locally controlled survival and OS will be performed in the per protocol population, which comprises all eligible patients who started their allocated treatment (at least one radiotherapy fraction as randomized). Sensitivity analyses according to intention-to-treat will include all patients as randomized. Patients free from progression and alive at the last visit will be censored for PFS at the day of last assessment. Patients free from in-field and margin progression and alive at the last visit will be censored for locally controlled survival at the day of last assessment. Patients not known to have died during the study will be censored for OS at the day they were last known to be alive. The primary comparison of the time-to-event distributions between the two treatment arms will be done using the one-sided log-rank test at significance level α = 10 % stratified by all factors used for randomization, to test H0: HR ≥ 1.0 versus H1: HR < 1.0. The HR for the experimental versus the control arm and its two-sided 80 and 95 % confidence intervals will be estimated using a Cox proportional hazards model stratified by the same factors. Distributions of PFS, locally controlled survival and OS will be estimated by the Kaplan-Meier method. The PFS and locally controlled survival rates at six months, the OS rates at one year, and medians of PFS, locally controlled survival and OS will be presented with two-sided 95 % confidence interval computed using the log-log transformation [34]. Additional exploratory analyses will study the prognostic impact of factors other than treatment, including age, sex and MGMT status.
Exploratory QoL analyses in the per protocol population will evaluate the evolution of QoL over time in the group of patients still alive at the respective time points. The results will be interpreted in conjunction with Kaplan-Meier estimates for OS. Cross-sectional descriptions of the average scores, which range from 0 to 100, will be presented by treatment arm with confidence intervals. Missing values will be imputed via linear regression models to assess the stability of the results. The primary analyses will classify the change scores according to the established minimal clinically important difference of 10 points [35] as (a) worsening vs unimportant worsening or improvement and (b) improvement vs unimportant improvement or worsening.
Analyses of the safety of FET application are performed in the pharmacovigilance population of all patients who received FET. Analyses of radiotherapy treatment toxicity are performed in the safety population of all patients who started treatment (at least one radiotherapy fraction), irrespective of eligibility, according to the treatment arm that they started. Rates of AEs occurring until incl. day 7 after FET-PET imaging and of SAEs occurring from start of radiotherapy until 30 days after end of radiotherapy will be presented with exact two-sided 95 % confidence intervals. Further analyses of treatment toxicity will present the worst grade of acute/subacute and late side effect by treatment arm. The time to occurrence of any severe (NCI-CTC grade ≥3) side effects and to any severe late side effects will be estimated by cumulative incidence. The time to severe (late) side effects will be calculated from the time of start of radiation treatment to the first evidence of any grade 3 (late) side effects. Patients alive without grade 3 (late) toxicity will be censored at the date of last follow-up, patients who died without experiencing (late) grade 3 side effects will be assessed as competing risk at the time of death.
Interim analyses
Initially, a first interim analysis comparing the GTV delineated according to FET uptake with the GTV delineated according to Gd enhancement in T1-weighted MRI was planned with the aim to stop the trial early for futility if the delineation would not show a difference in target volumes in a sufficient number of patients. However, in the meantime a monocenter feasibility study (German Clinical Trials Registry DRKS00000633) was started, and the protocol of the present multicenter trial was updated. It was decided that if the feasibility study would show relevant PET/MRI-GTV non-overlap in a substantial proportion of patients (> 25 %), the first interim analysis of the present multicenter trial would be cancelled. The results of the feasibility study have turned out as expected, so that the interim analysis of GTVs in the present trial will not be performed (unpublished data).
A second interim analysis is planned with the aim to stop the trial early for futility if the experimental arm shows no favourable trend in terms of the trial primary endpoint PFS. It will be performed in the per protocol population when the first 125 patients have been randomized, which was initially expected to occur at month 15 from study start. The test will be carried out as described in the paragraph on sample size. The trial will be stopped for futility (accept H0) if the p-value is above 0.51742.
Additionally, the rate of patients registered but not randomized to the study is regularly monitored descriptively during the recruitment phase. The aim is to assess the true feasibility of the study and to allow for corrective measures if there is a selection bias towards patients with large volumes or big differences in volumes not being randomized.
Discussion
The goal of this study is to evaluate if the delineation of the target volume based on AA-PET could have an impact on the clinical outcome in patients with recurrent gliomas treated with 3D-high conformal re-irradiation.
The selection of this disease is based on several considerations. First of all, treatment of recurrent GBM is an unsolved clinical problem and the prognosis of patients has remained poor despite intense research [36]. Thus, new treatment approaches including re-irradiation are urgently needed [37]. Second, local recurrence after surgery and radiotherapy is the cause of disease progression in almost all patients. Therefore, the impact of a local treatment approach with high precision radiotherapy on patient survival is expected to be higher than for other malignant diseases, which also or predominantly recur systemically [38]. Combined with the poor prognosis of recurrent GBM, this means that the impact of molecular imaging on patient outcome can be studied in a relatively small patient population and with a relatively short follow-up period. Third, in patients treated with re-irradiation the dose should be focused on the GTV, sparing the normal brain tissue as much as possible [14]. The treatment should be performed using high-precision radiation therapy, which is able to focus the dose on the macroscopic tumor mass and to spare the normal brain tissue [2]. The accuracy in the GTV definition should have a significant impact on local tumor control and should translate into improved clinical outcome [9]. Forth, there is considerable evidence that current approaches for target delineation in recurrent GBM are limited due to unspecific treatment related changes seen on CT and MRI [4]. Fifth, there is substantial data that PET imaging with radiolabeled amino acids provides more accurate information about tumor extension than MRI or CT, especially after pretreatment [39]. Sixth, the reported differences in tumor extension observed between MRI and AA-PET, the so-called non-overlap volume of the GTVs, are significant and appear robust enough to be tested in a multicenter clinical trial [8 and unpublished data from a monocentric feasibility study]. The magnitude of the differences between AA-PET and MRI also makes it clinically highly relevant to test AA-PET based radiation treatment planning in a clinical trial. Considering the reported high sensitivity and specificity of AA-PET for detection of recurrent GBM and the large differences in tumor extension between PET and MRI observed in patients scheduled to undergo radiotherapy, there appears to be a high risk that large parts of the tumor mass are missed when radiotherapy is based on the findings on MRI only. Finally, in preparation of this randomized multicenter trial, the principal investigators performed a small monocenter non-randomized study showing that survival is significantly improved when AA-PET is implemented into radiation treatment planning [9].
Taken together, considering that in patients treated with re-irradiation we will focus the dose on the GTV, using very small margins to CTV and PTV, we consider that this is a valid clinical model for demonstrating the differences between AA-PET and T1-Gd-MRI in the visualization of GTV, and the consequences on the clinical outcome. As outlined above, we believe that recurrent GBM represents an ideal model to study the impact of molecular imaging on patient management in a randomized clinical trial.
Furthermore, the lessons from this study could also be extrapolated to primary gliomas. The results could have a significant impact on tumor mass detection, treatment planning (surgery, radiation therapy, chemotherapy, immunotherapy, gene-virotherapy which needs to be directed towards active tumor areas, etc.). An accurate visualization of tumor mass will have a significant impact on the evaluation of tumor response after treatment and on treatment monitoring and could lead to a “modified Response Evaluation Criteria In Solid Tumors (RECIST)” or RANO definition adapted for PET imaging in neurooncology.
Therefore, we consider that the results of this study could change significantly the treatment strategy in brain tumors, if in addition confirmed in a phase III trial. Another important goal of this trial is to develop a strategy for GTV delineation based on PET. Considering the increasing impact of PET in radiation treatment planning, we consider that our trial will have a significant impact on the development of systematic strategies for tumor delineation based on PET. The lessons from this study will be generally useful for the development of a biological based treatment planning, also for other tumor entities. Additionally, in the context of the pharmacovigilance aspects of this study, the safety of FET application in clinical routine as AA-PET-tracer will be evaluated.
Abbreviations
- AA:
-
Amino acid
- CT:
-
Computed tomography
- CTV:
-
Clinical target volume
- EQD2 :
-
Equivalent dose in 2 Gy fractions
- FET:
-
O-(2-[18F]fluoroethyl)-L-tyrosine
- FU:
-
Follow up
- GBM:
-
Glioblastoma
- Gd:
-
Gadolinium
- GTV:
-
Gross tumor volume
- Gy:
-
Gray
- HR:
-
Hazard ratio
- MET:
-
L-[methyl-11C]methionine
- MGMT:
-
O-6-methylguanine-DNA methyltransferase
- MRI:
-
Magnetic resonance imaging
- OS:
-
Overall survival
- PET:
-
Positron emission tomography
- PFS:
-
Progression free survival
- PTV:
-
Planning target volume
- QoL:
-
Quality of life
- RANO:
-
Response Assessment in Neurooncology
- RECIST:
-
Response Evaluation Criteria In Solid Tumors
- RT:
-
Radiotherapy
- SAE:
-
Serious adverse event
- SFRT:
-
Sterotactic fractionated radiotherapy
- SPECT:
-
Single-photon emission computed tomography
- TMZ:
-
Temozolomide
References
Verellen D, De Ridder M, Linthout N, Tournel K, Soete G, Storme G. Innovations in image-guided radiotherapy. Nat Rev Cancer. 2007;7:949–60.
Burnet NG, Jena R, Burton KE, Tudor GS, Scaife JE, Harris F, Jefferies SJ. Clinical and practical considerations for the use of intensity-modulated radiotherapy and image guidance in neuro-oncology. Clin Oncol (R Coll Radiol). 2014;26:395–406.
Whitfield GA, Kennedy SR, Djoukhadar IK, Jackson A. Imaging and target volume delineation in glioma. Clin Oncol (R Coll Radiol). 2014;26:364–76.
Grosu AL, Weber WA. PET for radiation treatment planning of brain tumours. Radiother Oncol. 2010;96:325–7.
Niyazi M, Brada M, Chalmers AJ, Combs SE, Erridge SC, Fiorentino A, et al. ESTRO-ACROP guideline “target delineation of glioblastomas”. Radiother Oncol. 2016;118:35–42.
Grosu AL, Weber W, Feldmann HJ, Wuttke B, Bartenstein P, Gross MW, et al. First experience with I-123-alpha-methyl-tyrosine spect in the 3-D radiation treatment planning of brain gliomas. Int J Radiat Oncol Biol Phys. 2000;47:517–26.
Grosu AL, Feldmann H, Dick S, Dzewas B, Nieder C, Gumprecht H, et al. Implications of IMT-SPECT for postoperative radiotherapy planning in patients with gliomas. Int J Radiat Oncol Biol Phys. 2002;54:842–54.
Grosu AL, Weber WA, Riedel E, Jeremic B, Nieder C, Franz M, et al. L-(methyl-11C) methionine positron emission tomography for target delineation in resected high-grade gliomas before radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63:64–74.
Grosu AL, Weber WA, Franz M, Stärk S, Piert M, Thamm R, et al. Reirradiation of recurrent high-grade gliomas using amino acid PET (SPECT)/CT/MRI image fusion to determine gross tumor volume for stereotactic fractionated radiotherapy. Int J Radiat Oncol Biol Phys. 2005;63:511–9.
Munck Af Rosenschold P, Costa J, Engelholm SA, Lundemann MJ, Law I, Ohlhues L, Engelholm S. Impact of [18F]-fluoro-ethyl-tyrosine PET imaging on target definition for radiation therapy of high-grade glioma. Neuro-Oncology. 2015;17.
Brandsma D, van den Bent MJ. Pseudoprogression and pseudoresponse in the treatment of gliomas. Curr Opin Neurol. 2009;22:633–8.
Hutterer M, Hattingen E, Palm C, Proescholdt MA, Hau P. Current standards and new concepts in MRI and PET response assessment of antiangiogenic therapies in high-grade glioma patients. Neuro Oncol. 2015;17:784–800.
Huang RY, Rahman R, Ballman KV, Felten SJ, Anderson SK, Ellingson BM, et al. The Impact of T2/FLAIR Evaluation per RANO Criteria on Response Assessment of Recurrent Glioblastoma Patients Treated with Bevacizumab. Clin Cancer Res. 2016;22:575–81.
Mayer R, Sminia P. Reirradiation tolerance of the human brain. Int J Radiat Oncol Biol Phys. 2008;70:1350–60.
Lawrence YR, Li XA, el Naqa I, Hahn CA, Marks LB, Merchant TE, Dicker AP. Radiation dose-volume effects in the brain. Int J Radiat Oncol Biol Phys. 2010;76:S20–7.
Combs SE, Gutwein S, Thilmann C, Huber P, Debus J, Schulz-Ertner D. Stereotactically guided fractionated re-irradiation in recurrent glioblastoma multiforme. J Neurooncol. 2005;74:167–71.
Fogh SE, Andrews DW, Glass J, Curran W, Glass C, Champ C, et al. Hypofractionated stereotactic radiation therapy: an effective therapy for recurrent high-grade gliomas. J Clin Oncol. 2010;28:3048–53.
Thorwarth D. Functional imaging for radiotherapy treatment planning: current status and future directions-a review. Br J Radiol. 2015;88:20150056.
Weber WA, Grosu AL, Czernin J. Technology Insight: advances in molecular imaging and an appraisal of PET/CT scanning. Nat Clin Pract Oncol. 2008;5:160–70.
la Fougère C, Suchorska B, Bartenstein P, Kreth FW, Tonn JC. Molecular imaging of gliomas with PET: opportunities and limitations. Neuro Oncol. 2011;13:806–19.
Plotkin M, Gneveckow U, Meier-Hauff K, Amthauer H, Feussner A, Denecke T, et al. 18F-FET PET for planning of thermotherapy using magnetic nanoparticles in recurrent glioblastoma. Int J Hyperthermia. 2006;22:319–25.
Miwa K, Matsuo M, Shinoda J, Oka N, Kato T, Okumura A, Ueda T, et al. Simultaneous integrated boost technique by helical tomotherapy for the treatment of glioblastoma multiforme with 11C-methionine PET: report of three cases. J Neurooncol. 2008;87:333–9.
Rickhey M, Koelbl O, Eilles C, Bogner L. A biologically adapted dose-escalation approach, demonstrated for 18F-FET-PET in brain tumors. Strahlenther Onkol. 2008;184:536–42.
Lee IH, Piert M, Gomez-Hassan D, Junck L, Rogers L, Hayman J, et al. Association of 11C-methionine PET uptake with site of failure after concurrent temozolomide and radiation for primary glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2009;73:479–85.
Vees H, Senthamizhchelvan S, Miralbell R, Weber DC, Ratib O, Zaidi H. Assessment of various strategies for 18F-FET PET-guided delineation of target volumes in high-grade glioma patients. Eur J Nucl Med Mol Imaging. 2009;36:182–93.
Weber DC, Casanova N, Zilli T, Buchegger F, Rouzaud M, Nouet P, et al. Recurrence pattern after [(18)F]fluoroethyltyrosine-positron emission tomography-guided radiotherapy for high-grade glioma: a prospective study. Radiother Oncol. 2009;93:586–92.
Niyazi M, Geisler J, Siefert A, Schwarz SB, Ganswindt U, Garny S, et al. FET-PET for malignant glioma treatment planning. Radiother Oncol. 2011;99:44–8.
Piroth MD, Pinkawa M, Holy R, Klotz J, Schaar S, Stoffels G, et al. Integrated boost IMRT with FET-PET-adapted local dose escalation in glioblastomas. Results of a prospective phase II study. Strahlenther Onkol. 2012;188:334–9.
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–72.
Groenvold M, Petersen MA, Aaronson NK, Arraras JI, Blazeby JM, Bottomley A, et al. The development of the EORTC QLQ-C15-PAL: a shortened questionnaire for cancer patients in palliative care. Eur J Cancer. 2006;42:55–64.
Nieder C, Astner ST, Mehta MP, Grosu AL, Molls M. Improvement, clinical course, and quality of life after palliative radiotherapy for recurrent glioblastoma. Am J Clin Oncol. 2008;31:300–5.
Korn EL, Arbuck SG, Pluda JM, Simon R, Kaplan RS, Christian MC. Clinical trial designs for cytostatic agents: are new approaches needed? J Clin Oncol. 2001;19:265–72.
Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31:103–15.
Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. USA: John Wiley & Sons; 1980.
Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16:139–44.
Weller M, Wick W. Neuro-oncology in 2013: improving outcome in newly diagnosed malignant glioma. Nat Rev Neurol. 2014;10:68–70.
Seystahl K, Wick W, Weller M. Therapeutic options in recurrent glioblastoma - An update. Crit Rev Oncol Hematol. 2016;99:389–408.
Oppitz U, Maessen D, Zunterer H, Richter S, Flentje M. 3D-recurrence-patterns of glioblastomas after CT-planned postoperative irradiation. Radiother Oncol. 1999;53:53–7.
Galldiks N, Langen KJ, Pope WB. From the clinician’s point of view - What is the status quo of positron emission tomography in patients with brain tumors? Neuro Oncol. 2015;17:1434–44.
Acknowledgements
We gratefully acknowledge the German Neurooncological Network (Neuroonkologische Arbeitsgemeinschaft) and Working Group Radiation Oncology (Arbeitsgemeinschaft Radiologische Onkologie) of the German Cancer Society (Deutsche Krebsgesellschaft, DKG) for their support. We thank the DMC members (Prof. Dr. Cordula Petersen, Prof. Dr. Jörg Kotzerke, Dr. Jochem König and Dr. Andrea Schaefer-Schuler) for their advisory opinion.
Funding
The GLIAA trial is supported by the Deutsche Krebshilfe e.V. (Grant No. 109511). This funding source had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Availability of data and materials
Not applicable.
Authors’ contributions
OO, TSJ, IG are assistant medical trial coordinators. UN is medical coordinator, member of the protocol committee, and deputy of the coordinating investigator (Radiation Oncology). MM is member of the protocol committee and deputy of the coordinating investigator (Nuclear Medicine). EG is member of the protocol committee and responsible for statistical planning and analysis. SSF is project coordinator (Clinical Trials Unit). IM is the central referencing neuroradiologist and member of the protocol committee. AW, BGB, SCS, WAW are members of the protocol committee and co-designed the trial. PTM is co-coordinating investigator (Nuclear Medicine) and member of the protocol committee. ALG is the principal investigator, coordinating investigator (Radiation Oncology), designed the study and conducts it. All authors contributed to the writing of the manuscript and read and approved the final version.
Authors’ information
The authors choose not to include more information.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Written informed consent to publish has been obtained from the person whose images were used in Fig. 2.

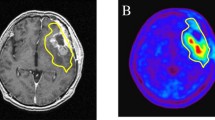
a and b Definition of GTV according to contrast enhancement in T1-MRI (green) and increased FET uptake (Tumor to Background Ratio >1.8, red). c and d Resulting PTV according to study arm A (FET-PET, pink). e and f Resulting PTV according to treatment arm B (MRI, pink). The corresponding treatment plan according to Arm A is shown in (g), and the corresponding treatment plan for Arm B is shown in (h). Isodose distribution is displayed as follows: 95 % isodose line (yellow), 80 % isodose line (green), and 50 % isodose line (blue). Source and copyright: Center for Diagnostic and Therapeutic Radiology, Medical Center – University of Freiburg, Faculty of Medicine, University of Freiburg, Germany
Ethics approval and consent to participate
The GLIAA trial has been approved by the Ethics Committee, Medical Center – University of Freiburg, Faculty of Medicine, University of Freiburg, Germany (No. 486/12, date of approval: 24.04.2014). Informed consent is obtained from all participants of the trial.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Oehlke, O., Mix, M., Graf, E. et al. Amino-acid PET versus MRI guided re-irradiation in patients with recurrent glioblastoma multiforme (GLIAA) – protocol of a randomized phase II trial (NOA 10/ARO 2013-1). BMC Cancer 16, 769 (2016). https://doi.org/10.1186/s12885-016-2806-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2806-z