
Abstract
Background
Novel therapies are needed for children with high-risk and relapsed neuroblastoma. We hypothesized that MAPK/ERK kinase (MEK) inhibition with the novel MEK1/2 inhibitor binimetinib would be effective in neuroblastoma preclinical models.
Methods
Levels of total and phosphorylated MEK and extracellular signal-regulated kinase (ERK) were examined in primary neuroblastoma tumor samples and in neuroblastoma cell lines by Western blot. A panel of established neuroblastoma tumor cell lines was treated with increasing concentrations of binimetinib, and their viability was determined using MTT assays. Western blot analyses were performed to examine changes in total and phosphorylated MEK and ERK and to measure apoptosis in neuroblastoma tumor cells after binimetinib treatment. NF1 protein levels in neuroblastoma cell lines were determined using Western blot assays. Gene expression of NF1 and MEK1 was examined in relationship to neuroblastoma patient outcomes.
Results
Both primary neuroblastoma tumor samples and cell lines showed detectable levels of total and phosphorylated MEK and ERK. IC50 values for cells sensitive to binimetinib ranged from 8 nM to 1.16 μM, while resistant cells did not demonstrate any significant reduction in cell viability with doses exceeding 15 μM. Sensitive cells showed higher endogenous expression of phosphorylated MEK and ERK. Gene expression of NF1, but not MEK1, correlated with patient outcomes in neuroblastoma, and NF1 protein expression also correlated with responses to binimetinib.
Conclusions
Neuroblastoma tumor cells show a range of sensitivities to the novel MEK inhibitor binimetinib. In response to binimetinib, sensitive cells demonstrated complete loss of phosphorylated ERK, while resistant cells demonstrated either incomplete loss of ERK phosphorylation or minimal effects on MEK phosphorylation, suggesting alternative mechanisms of resistance. NF1 protein expression correlated with responses to binimetinib, supporting the use of NF1 as a biomarker to identify patients that may respond to MEK inhibition. MEK inhibition therefore represents a potential new therapeutic strategy for neuroblastoma.
Similar content being viewed by others
Background
Neuroblastoma is the most common extracranial solid tumor in children, and patients with high-risk disease have very poor outcomes, with long term disease-free survival rates between 35 and 45 % despite aggressive treatment regimens [1–3]. High-risk cases are characterized by frequent relapses and tumors resistant to established treatment, and novel therapies are sorely needed for patients with high-risk and relapsed neuroblastoma. Since aberrant growth factor receptor expression and activity have been shown to contribute to neuroblastoma pathogenesis, downstream intracellular signaling pathways, including the RAS/mitogen-activated protein kinase (MAPK) pathway, represent potential therapeutic targets.
The RAS/MAPK signaling pathway is one of the most frequently dysregulated signaling cascades in human cancer. In the canonical pathway, activity of the small GTPase RAS leads to sequential phosphorylation and activation of three protein kinases, BRAF, MAPK/extracellular signal-regulated kinase (ERK) kinase 1/2 (MEK1/2), and extracellular signal-regulated kinase 1/2 (ERK1/2) [4, 5]. Physiological activation of MEK1/2 and ERK1/2 is required for multiple normal cellular processes; however, overactivation of the pathway can lead to malignant transformation. Both MEK1 and MEK2 represent potential targets for therapeutic development due to their homology, narrow substrate specificities, and unique structural characteristics.
Targeting MEK1/2 to inhibit the oncogenic activity of the RAS/MAPK signaling pathway has been shown to be effective in in vitro and in vivo preclinical studies [6–11]. Inhibitor binding to the MEK1/2 proteins leads to conformational changes that lock unphosphorylated MEK1/2 into catalytically inactive states [12–14]. Since this inhibitor binding site is separate from the ATP-binding site, the mechanism of inhibition is independent of ATP and, thus, off-target effects are largely avoided [14, 15]. Such studies have led to the development of more than a dozen small-molecule inhibitors of MEK. Binimetinib is an ATP-noncompetitive inhibitor of both MEK1 and MEK2. Initial in vitro kinase assays demonstrated MEK inhibition with an IC50 of 12 nM without inhibition of other kinases at doses up to 10 μM [16, 17], and the safety and pharmacokinetics of binimetinib have been evaluated in adult cancer patients in multiple phase I and II studies [18–26].
The role of the RAS/MAPK pathway in neuroblastoma pathogenesis is poorly understood. Activating mutations in the genes of members of the RAS-MAPK pathway have been identified in a small subset of neuroblastoma tumors at diagnosis [27] and in many neuroblastoma tumors after relapse [28]. Furthermore, recent studies have identified a potential role for the Ras-GTPase activating protein (RasGAP) NF1 as a mediator of CRA resistance in neuroblastoma cells [29], suggesting key roles for the RAS/MAPK pathway both in neuroblastoma differentiation and relapse. Based on the evidence for a role of RAS/MAPK signaling in oncogenesis, we hypothesized that binimetinib may show significant antitumor activity in preclinical studies of neuroblastoma.
Methods
Cells and culture conditions
The neuroblastoma cell lines used in this study have been previously described [30–38] and were generously provided by Shahab Asgharzadeh (Children’s Hospital Los Angeles, Los Angeles, CA), Susan Cohn (The University of Chicago Children’s Hospital, Chicago, IL), Jill Lahti (St. Jude Children’s Research Hospital, Memphis, TN), John Maris (Children’s Hospital of Philadelphia, Philadelphia, PA), William Weiss (The University of California, San Francisco, San Francisco, CA) or were purchased from the American Type Culture Collection (ATCC; Rockville, MD). Cell lines were grown at 37° in 5 % CO2 in appropriate media (Invitrogen, Carlsbad, CA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS) (Life Technologies, Grand Island, NY), L-glutamine, sodium pyruvate, and non-essential amino acids [39]. All cell lines were authenticated by deoxyribonucleic acid (DNA) profiling prior to use.
Patient-derived tumor samples
The patient tumor samples employed in these studies were obtained from the Texas Children’s Hospital Research Tissue Support Services tissue bank. Fresh, resected neuroblastoma tumor samples were collected from patients after informed consent from either the patients or their guardians was obtained via an Institutional Review Board-approved tissue banking protocol. Samples were placed in sterile human stem cell media at the time of collection and flash frozen in liquid nitrogen for storage. All experiments on patient tissue samples were performed in compliance with the Helsinki Declaration and were approved by the Baylor College of Medicine Institutional Review Board (H-29553).
Therapeutic agents
Binimetinib was generously provided by Novartis, Inc.. A 10 mM stock solution was generated in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO) and stored at −20 °C. Binimetinib was diluted in PBS or appropriate media immediately before use.
RAS/MAPK assays
Patient tumor samples were homogenized and incubated for 30 min in radioimmunoprecipitation assay (RIPA) protein lysis buffer containing protease inhibitors (Sigma) and phosphatase inhibitors (Roche, San Francisco, CA) with homogenization every 10 min as previously described [39]. Lysates were centrifuged and supernatants were collected. Neuroblastoma cells were plated in 100-mm plates and allowed to adhere and proliferate for 48 h. Media was replaced 24 h after plating. Cells from plates at approximately 80 % confluency were then harvested and lysed as above.
To measure the effects of binimetinib on MEK and ERK phosphorylation, 2 × 106 neuroblastoma cells were plated in 60-mm plates and allowed to adhere and proliferate for 48 h. Media was replaced 24 h after plating. Cells were treated with either 1 μM binimetinib or media alone (vehicle treatment) for one hour. Cells were harvested and lysed as above at the completion of each experiment.
Protein concentration in each sample lysate was measured using a protein assay dye reagent (Bio-Rad, Hercules, CA). 30–50 μg total denatured protein from each cell line or tumor sample lysate was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose or polyvinylidene fluoride (PVDF) membranes (Invitrogen, Carlsbad, CA) using standard techniques. Membranes were blocked in Odyssey blocking buffer (Li-Cor, Lincoln, NE) for two hours at room temperature and then incubated overnight with primary antibodies to total MEK (9126; 1:1000; Cell Signaling, Danvers, MA), phosphorylated MEK (9154; 1:1000; Cell Signaling), total ERK (4695; 1:1000; Cell Signaling), phosphorylated ERK (4370; 1:2000; Cell Signaling), NF1 (sc-67; 1:50; Santa Cruz Biotechnology), Actin (A5316 or A5441; 1:5000; Sigma), or Vinculin (1:10000; ab1290002; Abcam). Bound primary antibodies were incubated for two hours at room temperature with IRDye800 conjugated affinity purified anti-rabbit or anti-mouse secondary antibodies (1:5000; Rockland, Gilbertsville, PA), and the signal was visualized using an Odyssey infrared imaging system (Li-Cor). Immunoblot band densities were determined with ImageJ (v1.46r, NIH) as previously described [39]. Relative intensity levels were determined by dividing the band intensity of the total protein by the intensity of the loading control protein and by dividing the intensity of the phosphorylated protein by the intensity of the total protein.
Cell viability assays
The viability of cells exposed to binimetinib was determined using a modified methyl tetrazolium (MTT; Sigma) assay as previously described [39]. 0.35–0.9 × 105 cells/ml of exponentially growing cells were plated in wells of 96-well plates. 24 h later, binimetinib was added to each well at specified concentrations, and the plates were incubated at 37 °C. 24, 48, 72, 96, or 120 h later, MTT was added to each well and plates were incubated at 37 °C for four h to allow for reduction of MTT to its insoluble formazan by remaining viable cells. Medium was aspirated and 150 μl of DMSO was added to each well to solubilize precipitated MTT. The optical density (OD) was immediately measured at 550 nm using a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Relative cell viability was calculated by subtracting the background OD of media alone and then dividing by the OD of control wells. Replicates of six wells were used for each drug concentration and assays were duplicated on separate days. IC50 values were derived using best-fit trendlines as previously described [39].
To determine cell appearance before and after treatment with binimetinib, cells were plated as above and treated with either 1 μM or 10 μM binimetinib for 72 h. Cells were visualized using an inverted microscope (Nikon Eclipse TE-300, Nikon, Tokyo, Japan) and images were acquired on an RS Photometrics CoolSNAP color digital camera (Roper Scientific) using RS Photometrics Image Software Version 1.9.2 (Roper Scientific).
Apoptosis assays
For assays to measure induction of apoptosis, 2 × 106 neuroblastoma cells were plated in 60-mm plates and allowed to adhere and proliferate for 24 h. Cells were then treated with either 1 μM binimetinib, 10 μM binimetinib, or media alone (vehicle treatment) for six or eight hours (CHP-212 cells), 96 or 120 h (SJ-NB-10 cells), or 120 h only (CHP-134, NGP cells). Cells were harvested and lysed at the completion of each experiment as described above. Thirty please use mg (with symbol for "micro") total denatured protein from each cell line was separated by SDS-PAGE and transferred to nitrocellulose membranes (Invitrogen) as above. Western blots were performed as described above using primary antibodies to Poly(ADP-ribose) polymerase (PARP; 1:500, 9542, Cell Signaling) or Vinculin (1:10000; ab1290002; Abcam), anti-rabbit secondary antibody (1:5000; Rockland, Gilbertsville, PA), and the Odyssey infrared imaging system (Li-Cor).
Analysis of patient outcomes compared to NF1 and MEK1 expression
We obtained microarray analysis results of neuroblastoma patient tumor samples from the National Cancer Institute (NCI) Oncogenomics Data Center Section (available at: http://pob.abcc.ncicrf.gov/cgi-bin/JK) from the databases “Neuroblastoma Prognosis Database,” “Neuroblastoma Prognosis Database-Oberthuer Lab,” and “Exon Array Neuroblastoma Database” as previously described [40]. All available patient data from these databases was included in our analysis. Using gene expression results from these databases, patients were divided into high and low NF1 and MEK1 gene expression groups by median-centered log2 ratios as detailed on the NCI Oncogenomics database website. Kaplan-Meier survival curves were plotted using the open-source statistical packages in R (R Foundation for Statistical Computing, Vienna, Austria; available at: http://www.r-project.org). We compared survival curves between the NF1 and MEK1 gene expression groups using log-rank tests to examine the association between expression and patient survival outcomes in the whole cohort and in patients with stage 4 neuroblastoma and in those with stage 1, 2, 3, or 4S neuroblastoma.
We obtained additional microarray analysis results of neuroblastoma patient tumor samples from the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl) using the Versteeg database. MEK1 and MEK2 probesets in each database with the highest average signals were selected for analysis. Kaplan-Meier analyses were performed online and the resulting survival curves and p values (obtained via the log-rank test) were downloaded as previously described [41].
Results
Neuroblastoma patient samples and tumor cell lines demonstrate RAS/MAPK pathway expression and activity
To examine the expression of components of the RAS/MAPK signaling pathway in neuroblastoma tumors, a cohort of patient tumor samples was analyzed by Western blot for total and phosphorylated MEK and ERK. Patient tumor samples showed a range of expression of total and phosphorylated components of this pathway (Fig. 1a, c). Neuroblastoma cell lines also showed varying levels of total and phosphorylated MEK and ERK (Fig. 1b, d). Although there was no apparent correlation between levels of phosphorylated MEK and phosphorylated ERK in these samples and cell lines, detectable levels of both phosphorylated MEK and ERK suggested activity of this pathway in neuroblastoma tumor cells and also suggested the potential efficacy of MEK inhibitors in neuroblastoma preclinical models.

Neuroblastoma patient samples and cell lines show expression and activity of components of the RAS/MAPK signaling pathway. a Neuroblastoma patient samples were lysed and Western blots for total MEK, phosphorylated MEK (p-MEK), total ERK, and phosphorylated ERK (p-ERK) were performed. Vinculin was used as a loading control. b A panel of nine neuroblastoma cell lines, HeLa cells and 293T cells were lysed and Western blots for total MEK, p-MEK, total ERK, and p-ERK were performed. Actin and vinculin were used as loading controls. c, d Relative MEK, p-MEK, ERK, and p-ERK western blot band intensities were determined and plotted for each tested tumor sample and cell line
Neuroblastoma tumor cell responses to binimetinib
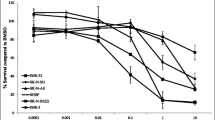
With the demonstrated activity of the RAS/MAPK pathway in neuroblastoma tumor cells and tumors, we hypothesized that MEK inhibition would lead to decreased cell viability. To investigate this hypothesis, neuroblastoma tumor cell lines were tested for sensitivity in vitro to the novel MEK1/2 inhibitor binimetinib. Four cell lines were sensitive to binimetinib and reached <50 % viability after 24 to 120 h of treatment (Fig. 2a–d) while five cell lines were resistant to the drug (Fig. 2e). Resistant cell lines were largely unaffected by treatment with binimetinib for up to five days with doses up to 15 μM (Fig. 2e, Additional file 1), while IC50 values for the sensitive cell lines ranged from 8 nM to 1.16 μM after 120 h of drug treatment (Fig. 2f). Resistant cell lines did not demonstrate any significant morphological changes in response to binimetinib, while sensitive cell lines demonstrated cell rounding and detachment from the surface, consistent with cell death (Additional file 2). Responsiveness of cells to binimetinib correlated with their levels of RAS/MAPK signaling pathway activity. Cell lines more sensitive to binimetinib tended to show higher levels of phosphorylated MEK and ERK proteins (Fig. 2g), while cell lines least sensitive to binimetinib showed lower levels of phosphorylated MEK and ERK proteins (Fig. 2g).

Neuroblastoma cell lines show bimodal responses to treatment with the MEK1/2 inhibitor binimetinib. a-f Neuroblastoma cells were treated with increasing concentrations of binimetinib for 24, 48, 72, 96, or 120 h and cell viability was determined by MTT assays. CHP-212 (log scale) (a), SK-N-BE(2) (b), SK-N-AS (c), and SJ-NB-10 (d) cells are sensitive to binimetinib treatment; e CHP-134, Kelly, LAN-5, NGP, and SK-N-DZ cells maintain resistance to binimetinib treatment after 120 h of drug exposure. f IC50 values (μM) were calculated for cells treated with binimetinib for 120 h. g Densitometry analysis was performed on Western blots from Fig. 1b to quantify relative phospho-ERK (pERK/ERK) protein levels in neuroblastoma tumor cell lines sensitive to binimetinib (“sensitive”) or resistant to binimetinib (“resistant”) h CHP-212 cells were treated with 1 μM binimetinib for 6 h (left two lanes) or 8 h (right two lanes) and SJ-NB-10 cells were treated with 1 μM binimetinib for 96 h (left two lanes) or 120 h (right two lanes). CHP-134 and NGP cells were treated with 1 μM or 10 μM binimetinib for 120 h. Cells were then lysed and Western blots for total and cleaved PARP were performed. Vinculin was used as a loading control
In order to determine the mechanism of decreased neuroblastoma tumor cell viability after treatment with binimetinib, we analyzed cells for cleavage of PARP before and after treatment with binimetinib. Treatment with binimetinib led to an increase in PARP cleavage in sensitive but not resistant cell lines (Fig. 2h), indicating that the reduction in viability from binimetinib treatment is at least partially due to induction of apoptosis in sensitive cell lines.
Binimetinib inhibits RAS/MAPK pathway activity
In order to demonstrate inhibition of MEK and ERK in neuroblastoma tumor cells, neuroblastoma tumor cell lines were treated with binimetinib or media alone for 1 h. Treatment of sensitive cell lines with binimetinib led to increased MEK phosphorylation and inhibition of ERK phosphorylation without changes in total levels of MEK and ERK protein (Fig. 3a, b). Resistant cell lines demonstrated either less robust increases in phosphorylation of MEK or incomplete inhibition of phosphorylated ERK (Fig. 3b), suggesting multiple possible mechanisms of resistance.

Binimetinib inhibits RAS/MAPK pathway activity. Neuroblastoma cells were treated with 1 μM binimetinib for 1 h and then lysed and Western blots for total MEK, phospho-MEK (p-MEK), total ERK, and phospho-ERK (p-ERK) were performed. Actin was used as a loading control
NF1 expression correlates with responses of cells to binimetinib
Expression of the RAS-GTPase activating protein (GAP) protein NF1 is associated with activity of the RAS/MAPK pathway, and mutations in or deletions of the NF1 gene have been found in a number of cancers, including neuroblastoma [29, 42]. To evaluate whether gene expression of RAS/MAPK pathway members was associated with neuroblastoma patient outcomes, we evaluated the associations of NF1 and MEK1/2 gene expression with neuroblastoma patient outcomes using results from microarray analyses of neuroblastoma tumors. NF1 gene expression, but not MEK1 or MEK2 gene expression, was strongly associated with patient outcomes in neuroblastoma and appeared to have prognostic effects independent of tumor stage (Fig. 4; Additional file 3).

Outcomes of patients with neuroblastoma based on MEK1 and NF1 gene expression. The NCI Oncogenomics gene expression databases were evaluated for outcomes of patients with neuroblastoma and Kaplan-Meier survival curves were generated. a Estimated overall survival for patients who have tumors with high NF1 gene expression (n = 177; gray) and low NF1 gene expression (n = 176; black) (log-rank test; p = 1.88e-11). b Estimated overall survival for patients with stage 4 neuroblastoma who have high (n = 35; black) and low (n = 90; dashed black) NF1 gene expression and for patients with tumors of all other stages with high (n = 142; gray) and low (n = 86; dashed gray) NF1 gene expression. c Estimated overall survival for patients who have tumors with high MEK1 gene expression (n = 154; gray) and low MEK1 gene expression (n = 153; black) (log-rank test; p = 0.44). d Estimated overall survival for patients with stage 4 neuroblastoma who have high (n = 44; black) and low (n = 54; dashed black) MEK1 gene expression and for patients with tumors of all other stages with high (n = 110; gray) and low (n = 99; dashed gray) MEK1 gene expression
Loss of or reduced expression of NF1 leads to hyperactivation of RAS and of its downstream signaling components such as MEK and ERK [43–45]. Thus, we hypothesized that NF1 expression in neuroblastoma tumor cells might influence responses to binimetinib treatment. To determine whether NF1 protein levels correlated with responses to binimetinib, we examined levels of NF1 protein in neuroblastoma cell lines. Cell lines sensitive to binimetinib treatment had the lowest NF1 protein levels, while resistant cell lines showed the highest levels of NF1 protein (Fig. 5), suggesting that NF1 levels may be useful as a biomarker to identify neuroblastoma patients that would be more likely to respond to MEK inhibitor therapy.

Neuroblastoma cell lines sensitive to binimetinib show lower levels of NF1 protein expression. a A panel of neuroblastoma cell lines was analyzed by Western blot for NF1 protein expression levels. Actin was used as a loading control. b Densitometry analysis was performed to quantify relative NF1 protein levels in neuroblastoma tumor cell lines sensitive or resistant to binimetinib
Discussion
New treatment strategies are sorely needed for patients with high risk and relapsed neuroblastoma, and we have shown that MEK inhibition with binimetinib may represent an effective therapy for these patients. We have shown that neuroblastoma tumor cells and patient samples show expression and activity of components of the RAS/MAPK signaling pathway, supporting a role for this pathway in neuroblastoma pathogenesis. We have also shown that multiple neuroblastoma tumor cell lines were sensitive to treatment with the MEK inhibitor binimetinib, with sensitivity to MEK inhibition linked to NF1 protein expression and levels of phosphorylated MEK and ERK. Furthermore, we have shown that NF1 gene expression is associated with neuroblastoma patient outcomes, suggesting that MEK inhibitors would be most effective in patients with the worst outcomes and that NF1 expression represents a potentially useful biomarker for response to RAS/MAPK pathway inhibition.
GTPase-activating proteins (GAPs), including NF1, function as negative regulators of RAS. RAS cycles between an active, GTP-bound state and an inactive, GDP-bound conformation. The interaction between RAS and NF1 accelerates the conversion of RAS-GTP to RAS-GDP, therefore downregulating the activity of RAS, and loss of NF1 leads to hyperactivation of RAS and of its downstream signaling components such as MEK and ERK [43–45]. Previous work has identified NF1 gene deletions in multiple neuroblastoma cell lines [29], likely contributing to the lack of NF1 protein seen in these cell lines and suggesting that neuroblastoma tumors with reduced or absent NF1 expression are likely to be sensitive to MEK inhibition.
Upon treatment with binimetinib, neuroblastoma tumor cells show a bimodal response with some cells being very sensitive and others being resistant. Our data indicates that binimetinib strongly suppresses ERK activity in the sensitive cell lines, leading to apoptosis. However, multiple neuroblastoma tumor cells are resistant to inhibition of MEK with binimetinib, with either incomplete inhibition of ERK phosphorylation or reduced increases in MEK phosphorylation after treatment. Therefore, mechanisms of resistance could include alternative signaling pathways or feedback loops activating ERK in the absence of MEK activity, leading to resistance to binimetinib. Research is ongoing to identify these pathways mediating resistance to MEK inhibitor therapy.
Binimetinib treatment in adult cancer patients was generally well-tolerated but was associated with mild to moderate central serous-like retinopathy, diarrhea and acneiform dermatitis, similar to other MEK inhibitors [25, 26, 46]. Currently, multiple clinical trials examining the safety and efficacy of binimetinib alone and in conjunction with other drugs for cancer therapy are ongoing. MEK inhibition with binimetinib also results in the inhibition of other normal physiologic processes, such as inflammation [16, 47]. Therefore, it will be crucial to identify patient subpopulations most likely to benefit from MEK inhibitor therapy to minimize the risk:benefit ratio for patients.
Conclusions
Currently, more than a dozen inhibitors of MEK1 and MEK2 are in clinical development, including binimetinib. In clinical trials, such inhibitors have shown a range of efficacy. Unfortunately, in many cases, patients fail to initially respond to treatment; in other cases, patients respond well at the onset of treatment but later develop mechanisms of resistance to such drugs. Being able to identify an appropriate patient population that will respond to inhibition of MEK would facilitate the effective development and use of such inhibitors. Our data has supported a model in which levels of NF1 and phosphorylated MEK and ERK influence the sensitivity of cell lines to MEK inhibition with binimetinib. However, levels of phosphorylated MEK and ERK would be difficult both to obtain and quantify, and therefore our data identifying NF1 as a biomarker that predicts both patient outcomes and responsiveness of neuroblastoma tumor cells to MEK inhibition supports a potential role for readily available genetic testing for NF1 mutations and deletions in tumor samples. Thus, NF1 may not only function as an easily obtainable prognostic marker to predict disease outcomes but NF1 gene and protein expression levels may also represent independent molecular markers for a subset of neuroblastoma patients that is in need of additional therapies and that may respond well to MEK inhibition.
Abbreviations
- ATCC:
-
American Type Culture Collection
- DMSO:
-
dimethyl sulfoxide
- ERK:
-
extracellular signal-regulated kinase
- FBS:
-
fetal bovine serum
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
MAPK/ERK kinase
- MTT:
-
3-(4,5 dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide
- NF1:
-
neurofibromatosis type 1
- PARP:
-
poly(ADP-ribose) polymerase
- PVDF:
-
polyvinylidene fluoride
- RIPA:
-
radioimmunoprecipitation assay
- SDS-PAGE:
-
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
References
Berthold F, Boos J, Burdach S, Erttmann R, Henze G, Hermann J, et al. Myeloablative Megatherapy with Autologous Stem-Cell Rescue Versus Oral Maintenance Chemotherapy as Consolidation Treatment in Patients with High-Risk Neuroblastoma: A Randomised Controlled Trial. Lancet Oncol. 2005;6:649–58.
Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol. 2009;27:1007–13.
Kreissman SG, Seeger RC, Matthay KK, London WB, Sposto R, Grupp SA, et al. Purged Versus Non-Purged Peripheral Blood Stem-Cell Transplantation for High-Risk Neuroblastoma (COG A3973): A Randomised Phase 3 Trial. Lancet Oncol. 2013;14:999–1008.
Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Op Cell Biol. 1997;9:180–6.
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40.
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci. 1995;92:7686–9.
Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6.
Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–19.
Daouti S, Higgins B, Kolinsky K, Packman K, Wang H, Rizzo C, et al. Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Mol Cancer Ther. 2010;9:134–44.
Winski S, Anderson D, Bouhana K, Rhodes S, Impastato R, Woessner R, et al. MEK162 (ARRY-162), a novel MEK1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. Eur J Cancer Suppl. 2010;8:56.
Bid HK, Kibler A, Phelps DA, Manap S, Xiao L, Lin J, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res. 2013;19:6716–29.
Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–74.
Liang H, Liu T, Chen F, Liu Z, Liu S. A full-length 3D structure for MAPK/ERK kinase 2 (MEK2). Sci China Life Sci. 2011;54:336–41.
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7.
Trujillo JI. MEK inhibitors: a patent review 2008–2010. Expert Opin Ther Pat. 2011;21:1045–69.
Pheneger J, Wallace E, Marlow A, Hurley B, Lyssikatos J, Bendele AM, Lee PA. Characterization of ARRY-438162, a potent MEK inhibitor in combination with methotrexate or ibuprofen in in vivo models of arthritis [abstract]. American College of Rheumatology; 2006 Annual Scientific Meeting.
Lee P, Wallace E, Marlow A, Yeh T, Marsh V, Anderson D, et al. Preclinical development of ARRY-162, a potent and selective MEK1/2 inhibitor [abstract]. In: Proceedings of the 101st Annual Meeting of the American Association for Cancer Research; 2010. Abstract number 2515.
Bendell JC, Papadopoulos K, Jones SF, Barrett E, Guthrie K, Kass CL, et al. Abstract B243: A phase I dose-escalation study of MEK inhibitor MEK162 (ARRY-438162) in patients with advanced solid tumors. Mol Cancer Ther. 2011;10(Supplement 1):B243.
Finn RS, Javie MM, Tan BR, Weekes CD, Bendell JC, Patnaik A, et al. A phase I study of MEK inhibitor MEK162 (ARRY-438162) in patients with biliary tract cancer [abstract]. J Clin Oncol. 2012;30(4_suppl):220.
Chi P, Qin LX, D’Angelo SP, Dickson MA, Gounder M, Keohan ML, et al. A phase Ib/II study of MEK162 (binimetinib) in combination with imatinib in patients with advanced gastrointestinal stromal tumor (GIST) [abstract]. J Clin Oncol. 2015;33(Suppl):10507.
Grisham RN, Gordon MS, Harb WA, Aghajanian C, McMeekin DS, McKinley K, et al. A phase Ib dose-escalation study of binimetinib (MEK162) in combination with weekly paclitaxel in patients with platinum-resistant epithelial ovarian, fallopian tube or primary peritoneal cancer [abstract]. J Clin Oncol. 2015;33(Suppl):5516.
Sosman JA, Kittaneh M, Lolkema MP, Postow M, Schwartz G, Franklin C, et al. A phase Ib/II study of LEE011 in combination with binimetinib (MEK162) in patients with advanced NRAS-mutant melanoma: early encouraging clinical activity [abstract]. J Clin Oncol. 2014;32(Suppl):9009.
Juric D, Soria JC, Sharma S, Banerji U, Azaro A, Desai J, et al. A phase Ib open-label, multicenter, dose-escalation and -expansion study of orally administered binimetinib (MEK162) plus BYL719 in adult patients with selected advanced solid tumors [abstract]. J Clin Oncol. 2014;32(Suppl):9051.
Shimokata T, Watanabe K, Shibata T, Inada-Inoue M, Shirao K, Hirashima Y, et al. Results of a phase 1 study of MEK162, an oral MEK inhibitor, in Japanese patients with advanced solid tumors [abstract]. Eur J Cancer. 2013;49:3746.
van Herpen CML, Agarwala SS, Hauschild A, Dummer R, Berking C, Beck JT, et al. Overall survival and biomarker results from a phase II study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS-mutant melanoma. Ann Oncol. 2014;25:1–41.
Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–56.
Shukla N, Ameur N, Yilmaz I, Nafa K, Lau C-Y, Marchetti A, et al. Oncogene Mutation Profiling of Pediatric Solid Tumors Reveals Significant Subsets of Embryonal Rhabdomyosarcoma and Neuroblastoma With Mutated Genes in Growth Signaling Pathways. Clin Cancer Res. 2011;18:748–57.
Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Gen. 2015;47:864–71.
Holzel M, Huang S, Koster J, Ora I, Lakeman A, Caron H, et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell. 2010;142:218–29.
Biedler JL, Helson L, Spengler BA. Morphology and Growth, Tumorigenicity, and Cytogenetics of Human Neuroblastoma Cells in Continuous Culture. Cancer Res. 1973;33:2643–52.
Brodeur GM, Green AA, Hayes FA, Williams KJ, Williams DL, Tsiatis AA. Cytogenetic Features of Human Neuroblastomas and Cell Lines. Cancer Res. 1981;41:4678–86.
Reynolds CP, Tomayko MM, Donner L, Helson L, Seeger RC, Triche TJ, et al. Biological Classification of Cell Lines Derived from Human Extra-Cranial Neural Tumors. Prog Clin Biol Res. 1988;271:291–306.
Schlesinger HR, Gerson JM, Moorhead PS, Maguire H, Hummeler K. Establishment and Characterization of Human Neuroblastoma Cell Line. Cancer Res. 1976;36:3094–100.
Sugimoto T, Tatsumi E, Kemshed JT, Helson L, Green AA, Minowada J. Determination of cell surface membrane antigens common to both human neuroblastoma and leukemia-lymphoma cell lines by a panel of 38 monoclonal antibodies. J Natl Cancer Inst. 1984;73:51–7.
Helson L, Helson C. Human neuroblastoma cells and 13-cis-retinoic acid. J Neurooncol. 1985;3:39–41.
Gilbert F, Feder M, Balaban G, Brangman D, Lurie DK, Podolsky R, et al. Human Neuroblastoma and Abnormalities of Chromosomes 1 and 17. Cancer Res. 1984;44:5444–9.
Shapiro D, Valentine M, Rowe S, Sinclair A, Sublett J, Roberts W, et al. Detection of N-myc gene amplification by fluorescence in situ hybridization; diagnostic utility for neuroblastoma. Am J Pathol. 1993;142:1339–46.
Schwab M, Alitalo K, Klempnauer K, Varmus H, Bishop J, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumor. Nature. 1983;305:245–8.
Zhang L, Scorsone K, Woodfield SE, Zage PE. Sensitivity of Neuroblastoma to the Novel Kinase Inhibitor Cabozantinib Is Mediated by ERK Inhibition. Cancer Chemother Pharmacol. 2015;76:977–87.
Zage PE, Sirisaengtaksin N, Liu Y, Gireud M, Brown BS, Palla S, et al. UBE4B levels are correlated with clinical outcomes in neuroblastoma patients and with altered neuroblastoma cell proliferation and sensitivity to epidermal growth factor receptor inhibitors. Cancer. 2013;119:915–23.
Fabian J, Lodrini M, Oehme I, Schier MC, Thole TM, Hielscher T, et al. GRHL1 Acts as Tumor Suppressor in Neuroblastoma and Is Negatively Regulated by MYCN and HDAC3. Cancer Res. 2014;74:2604–16.
The I, Murthy AE, Hannigan GE, Jacoby LB, Menon AG, Gusella JF, et al. Neurofibromatosis type 1 gene mutations in neuroblastoma. Nat Gen. 1993;3:62–6.
Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–5.
Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–9.
Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–9.
Urner-Bloch U, Urner M, Stieger P, Galliker N, Winterton N, Zubel A, et al. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann Oncol. 2014;25:1437–41.
Bhagwat SS. MAP Kinase Inhibitors in Inflammation and Autoimmune Disorders. Ann Rep Med Chem. 2007;42:265–78.
Acknowledgements
We would like to acknowledge Novartis, Inc. for providing study drug and research funding in support of this project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
This study was supported by research funding and study drug from Novartis, Inc. to P.E.Z. All other authors declare that they have no conflict of interest.
Authors’ contributions
SEW, LZ, and KAS carried out experiments for the study, and SEW compiled the results, generated the figures for the paper, and prepared the manuscript. YL analyzed the patient tumor expression data and generated the Kaplan-Meier curves. PEZ conceived the study, designed the experiments, directed the project and helped edit and submit the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
CHP-134, Kelly, LAN-5, NGP, and SK-N-DZ cells remain resistant to binimetinib at doses exceeding 15 μM. Neuroblastoma cells were treated with increasing concentrations of binimetinib for 120 h and cell viability was determined by MTT assays. (PPTX 60 kb)
Additional file 2:
Effects of binimetinib on neuroblastoma tumor cell morphology. Neuroblastoma tumor cells were photographed before treatment and after treatment with 1 μM or 10 μM binimetinib for 72 h. (PPTX 12467 kb)
Additional file 3:
Using the neuroblastoma Versteeg patient data-sets in the R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl), patients were divided into high (blue) and low (red) MEK1 (left) and MEK2 (right) gene expression groups by median-centered Log2 ratios and survival curves were generated. Overall survival curves are shown with patient numbers in parentheses. (PPTX 144 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Woodfield, S.E., Zhang, L., Scorsone, K.A. et al. Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 16, 172 (2016). https://doi.org/10.1186/s12885-016-2199-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2199-z