
Abstract
Background
Cachexia is a multi-factorial, systemic syndrome that especially affects patients with cancer of the gastrointestinal tract, and leads to reduced treatment response, survival and quality of life. The most important clinical feature of cachexia is the excessive wasting of skeletal muscle mass. Currently, an effective treatment is still lacking and the search for therapeutic targets continues. Even though a substantial number of animal studies have contributed to a better understanding of the underlying mechanisms of the loss of skeletal muscle mass, subsequent clinical trials of potential new drugs have not yet yielded any effective treatment for cancer cachexia. Therefore, we questioned to which degree findings from animal studies can be translated to humans in clinical practice and research.
Discussion
A substantial amount of animal studies on the molecular mechanisms of muscle wasting in cancer cachexia has been conducted in recent years. This extensive review of the literature showed that most of their observations could not be consistently reproduced in studies on human skeletal muscle samples. However, studies on human material are scarce and limited in patient numbers and homogeneity. Therefore, their results have to be interpreted critically.
Summary
More research is needed on human tissue samples to clarify the signaling pathways that lead to skeletal muscle loss, and to confirm pre-selected drug targets from animal models in clinical trials. In addition, improved diagnostic tools and standardized clinical criteria for cancer cachexia are needed to conduct standardized, randomized controlled trials of potential drug candidates in the future.
Similar content being viewed by others


Background
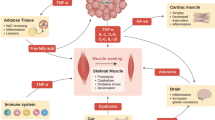
Cancer cachexia is a multi-factorial, systemic syndrome that occurs in the course of malignant diseases, especially in cancer of the gastrointestinal tract (GIT) [1, 2]. When all types of cancer are considered, cachexia affects around 60 % of patients in the course of their disease [3]. In gastric or pancreatic cancer even 80 % of patients are affected [1, 2, 4, 5]. In addition, cachexia is also observed in the course of benign diseases like chronic heart failure, renal failure and chronic obstructive pulmonary disease (COPD). The cachexia syndrome is characterized by weight loss due to excessive wasting of skeletal muscle and adipose tissue mass, which usually cannot be reversed by conventional nutritional support and is frequently accompanied by anorexia, fatigue, anemia and abnormal metabolism [2, 6, 7]. In cancer patients, cachexia can occur in every stage of disease and is associated with a poor prognosis, reduced treatment tolerance and a marked reduction in quality of life (QoL). The diagnostic criteria for cancer cachexia are weight loss >5 % or weight loss >2 % in individuals with a body mass index (BMI) <20 kg/m2 over 6 months in absence of simple starvation, or the presence of sarcopenia (skeletal muscle index <7.26 kg/m2 for males and <5.45 kg/m2 for females) with any degree of weight loss >2 % [8]. Furthermore, reduced food intake, anorexia, markers of systemic inflammation like C-reactive protein (CRP), responsiveness to chemotherapy and the rate of cancer progression should be assessed for the diagnosis of cancer cachexia [8]. The presence of cachexia in cancer patients is associated with reduced treatment response and tolerance and accounts for at least 20 % of cancer-specific mortality [2, 4, 9]. Furthermore, cachectic patients have an elevated surgical risk. Therefore, preservation of lean body mass can be critical for the survival of cancer patients, but an effective treatment for cachexia is still lacking.
Skeletal muscle wasting is the most important phenotypic feature of cancer cachexia and is among the principle causes of functional impairment, respiratory complications and fatigue [10, 11]. A recent study on cachectic patients with cancer of the GIT showed that besides the loss of muscle mass there is a substantial loss of muscle strength and mechanical quality [11]. In contrast to starvation, the non-muscle protein compartment of the body remains relatively unaffected and the liver mass is even increased, implying a tumor-associated metabolic condition that specifically targets skeletal muscle and subcutaneous adipose tissue [7]. Even though pathophysiological mechanisms of the depletion of skeletal muscle tissue during cancer cachexia have been intensely studied in recent years, identification of the key processes and therapeutic targets has been impeded by the large number of mediators and signaling pathways involved [12]. Furthermore, there is evidence of complex tissue interactions in this systemic syndrome, mediated through cytokines, tumor-derived factors, hormones and neuropeptides [1, 6, 12, 13]. However, so far the majority of studies have been conducted in animal models and studies of human muscle biopsies remain scarce and inconsistent [14]. The aim of this review is to outline these discrepancies and investigate to which degree findings from animal studies are translatable into clinical practice and research.
Mediators and signaling pathways in cancer cachexia – findings from animal models vs. human samples
Cancer cachexia is a complex systemic syndrome that involves a large number of systemic pro- and anti-inflammatory mediators as well as hormones, neuropeptides and tumor derived factors. However, in the following only those systemic mediators and pathways, on which sufficient comparable studies in animals as well as humans were available, will be first reviewed and then discussed. Table 1 gives an overview of these mediators and mechanisms and the findings regarding their role in animal vs. human studies.
Cytokines
TNF-α, TRAF6
Tumor necrosis factor (TNF)-α and TNFR-1 mRNA were shown to be elevated in several animal models of cancer cachexia and pharmacological inhibition of TNF-α showed a reduction in weight loss due to cancer in rodents [15–21]. As was recently reviewed by Baracos et al., TNF-α especially seems to play a role in the Yoshida hepatoma and sarcoma rat model as well as the Lewis lung carcinoma (LLC) model, but not in the C26- or MAC16- adenocarcinoma mouse models [22]. Recently, TNF-α receptor adaptor protein 6 (TRAF6) [23–26] which functions as a E3 ubiquitin ligase, has also been shown to be involved in catabolic signaling of cachexia in LLC mice [25].
In humans, several studies also found correlations of TNF-α serum levels with cachexia. A study on patients with pancreatic cancer showed that serum TNF-α levels were inversely correlated with BMI, hematocrit, hemoglobin, serum protein and albumin levels [27] and similar observations were made in patients with prostate cancer [28, 29] and hepatocellular carcinoma [30]. In addition, it was shown that expression of the TNF-α gene was upregulated in patients with pancreatic cancer and normalized after the tumor was surgically resected [31]. Others observed significant differences in serum TNF-α of patients and controls, but no correlation with weight loss [32, 33]. Interestingly, a recent study on 102 gastric cancer patients showed that TRAF6 mRNA and protein, as well as ubiquitin mRNA and protein, were all upregulated in skeletal muscle tissue and correlated with disease stage and the degree of weight loss. The positive correlation between TRAF6 and ubiquitin expression suggests that TRAF6 may regulate ubiquitin activity in human cancer cachexia [34].
Interleukin-6
Another pro-inflammatory mediator with a critical role in muscle wasting during cancer cachexia is interleukin (IL)-6 [35]. Elevated serum IL-6 levels have been observed in C26 and ApcMin/+ mouse models of cancer cachexia, and systematic administration of IL-6 to these mice resulted in depletion of skeletal muscle and adipose tissue and ultimately led to death. Furthermore, pharmaceutical inhibition of IL-6 signaling was shown to decrease the rate of cachexia in tumor-bearing rodents [35–39]. In skeletal muscle, the three most important intracellular signaling pathways induced by the ligand-receptor binding of IL-6 are the activation of JAK/STAT3, ERK and PI3K/Akt pathways [40–42]. In vitro tests have shown that the activation of STAT3 is both necessary and sufficient to induce muscle wasting. ApcMin/+ mice also showed increased activation of STAT3 in skeletal muscle [37]. Pharmacological inhibition of STAT3 was able to reduce muscle atrophy in mice with colon carcinoma; however, it was not sufficient to completely attenuate cachexia [40].
In human studies, elevated serum IL-6 levels were quite consistently associated with weight loss and a reduced rate of survival in cancer patients [1, 35, 40, 43–51]. Moreover, IL-6 was shown to be significantly over-expressed in pancreatic cancer tissue, and serum levels were significantly elevated in cachectic compared to non-cachectic patients with pancreatic cancer [48, 52, 53] and prostate cancer [49].
IL-1β and INF-γ
In some animal models, IL-1 and interferon (INF)-γ were shown to induce weight loss and anorexia, and neutralizing IFN-γ antibodies successfully attenuated cachexia [16]. In particular IL-1ß appears to be essentially involved in the central regulation of food intake and feeding behavior [54]. In a study of patients with advanced upper GIT cancer or NSCLC, IL-1ß was shown to be a better predictor of cachexia than IL-6, which did not correlate with weight loss in this study population [44]. In another study on GIT cancer patients, a correlation between weight loss and serum vascular endothelial growth factor (VEGF)-A, as well as between VEGF-A, IL-6 and IL-1 serum levels were observed [32, 33]. However, there are also several studies that did not find any correlation of serum cytokine levels with weight loss or cachexia in cancer patients [55–57].
Increased proteolysis
Myostatin
Mediators of increased proteolysis in cancer cachexia include members of the transforming growth factor (TGF)-β family. Myostatin is a secreted protein expressed predominantly in skeletal muscle and to a lesser extent in cardiac muscle and adipose tissue [58–60]. It has recently been shown that myostatin is also secreted by C26 carcinoma cells and other murine and human neoplasms [61]. Free myostatin binds to a high-affinity activin type-2 (ActRIIB) receptor in skeletal muscle, which induces a range of intracellular signaling cascades leading to increased proteolysis [4, 58, 59, 61, 62], as well as the inhibition of anabolic pathways (IGF-1/Akt) [63–65] and muscle regeneration [59, 61, 62, 66]. In a murine model of cancer cachexia, inhibition of myostatin by specific antibodies was able to attenuate the atrophy of skeletal muscle and improved muscle mass and function [67, 68]. In addition, blocking of the ActRIIB receptor attenuated the wasting process in skeletal muscle and heart and was associated with increased survival of tumor-bearing mice in several models of cancer cachexia [13, 58, 69]. Furthermore, this treatment brought elevated serum levels of myostatin back to normal and attenuated cachexia, independently of pro-inflammatory cytokine levels [70]. Other TGF-ß family members that act through the ActRIIB receptor are activin A, inhibin and macrophage inhibitory cytokine-1 (MIC-1/GDF-15) [58]. In mouse models, animals given either myostatin or activin A showed up to a 30 % decrease in muscle mass [13, 58, 71]. Furthermore, the inhibition of activin A was able to rescue myoblasts treated with TNF-α or IL-1 and allowed normal differentiation into myotubes [72]. In addition, activin A signaling has been linked to increased mitochondrial energy metabolism and oxygen consumption, supporting its role in maintaining body weight and resting energy expenditure (REE) [73]. Treatment with a specific anti-activin A antibody was able to prevent cachexia and death in a mouse model [13, 70]. Another member of the TGF-ß family, Inhibin is a secreted tumor suppressor and is a competitive antagonist of activin for the ActRIIB receptor. Inhibin deficiency causes gonadal tumor growth and severe cachexia in animals. Treatment of inhibin-deficient mice with an ActRIIB antibody prevented cachexia, reduced tumor growth, and prolonged survival [74, 75]. Moreover, it has recently been shown that MIC-1 over-expressing tumor-bearing mice showed decreased food intake and increased loss of muscle and fat mass. The degree of serum MIC-1 elevation directly correlated with the amount of cancer-related weight loss, which was reversed by neutralization of MIC-1 with a specific monoclonal antibody [76].
Studies investigating the relevance of myostatin or ActRIIB signaling in human cancer patients are scarce. Aversa et al. observed that myostatin was significantly increased in patients with gastric cancer who did not lose weight, whereas Smad2 expression was unchanged. Interestingly, in the same study, lung cancer patients had no increase in myostatin serum levels but did have increases in Smad2 expression, raising the question whether different tumors might induce different patterns of molecular changes within skeletal muscle tissue [77].
Proteolysis-inducing factor
A much-debated mediator of increased proteolysis and muscle loss in cancer cachexia is the tumor derived proteolysis-inducing factor (PIF). In experimental cancer cachexia, PIF has been shown to induce degradation of skeletal muscle protein via the ubiquitin proteasome system (UPS) [78–84]. Proteolysis-inducing factor was first isolated from the urine of tumor-bearing mice, and was shown to induce the increased expression of proteasome subunits and increased proteasome activity via NF-κB [82, 85–87]. The activation of NF-κB includes the phosphorylation of RNA-dependent protein kinase (PKR), which inhibits protein synthesis [78]. The relevance of this process to cancer cachexia is demonstrated by the ability of a PKR-inhibitor to abate skeletal muscle atrophy in a mouse model of cachexia by attenuating UPS-dependent proteolysis and increasing protein synthesis [88].
However, in human cancer cachexia, the existence of a homologue to PIF is controversial [86, 89]. First, a homologue glycoprotein was isolated from urine of weight-losing cancer patients, and when purified and injected into mice, it induced a 10 % loss of body weight within 24 h [54]. Subsequently, several studies showed an association between PIF and weight loss in patients with cancer cachexia [90–93]. Immunohistochemical staining of tumor samples from patients with GIT cancers showed that PIF was expressed by the tumor, which was strongly associated with weight loss and the presence of PIF in urine samples [91]. However, the only longitudinal study on the presence of PIF in urine of 36 GIT cancer patients showed that over time, cancer patients positive for the PIF pattern experienced weight loss, whereas those with a negative test gained weight [92]. Based on the available sequence of PIF, the factor HCAP (Human cachexia-associated protein) was identified in cell lines, in metastatic tumors and in the urine of cancer patients with cachexia [93]. In addition, the same research group showed that the expression of HCAP in prostate cancer cells was associated with disease progression and the development of cachexia [93]. In contrast, other groups found that the presence of PIF in urine did not correlate with weight loss, anorexia, tumor response, or survival in cancer patients [89, 94].
Angiotensin II
Similar to the action of PIF, angiotensin-II (AngII) has been shown to directly accelerate protein breakdown by the UPS in vitro [95]. In animals, it was shown that proteolytic system compounds were upregulated in skeletal muscle by AngII, and the subsequent increase in protein degradation was blocked by muscle-specific expression of IGF-1 [95–97]. The action of AngII involves activation of NF-κB-dependent signaling and the formation of reactive oxygen species (ROS) [98]. In mice with cancer cachexia, inhibition of ROS formation by the antioxidant α-tocopherol was able to rescue skeletal muscle mass [80, 99]. Furthermore, in a recent study on the effect of treatment of C26-mice with angiotensin-converting enzyme (ACE) inhibitors, it was shown that muscle mobility and strength, as well as respiratory function were improved although body and muscle mass were not increased [100].
However, the role of AngII in human cancer cachexia remains to be determined. In patients with cachexia related to congestive heart failure, treatment with ACE inhibitors caused an increase in both subcutaneous fat and muscle mass [101]. There is also some preliminary evidence that ACE inhibitors have the potential to ameliorate cancer cachexia, at least in NSCLC patients [102]. In addition, treatment with antioxidants has been shown to be effective in increasing lean body mass, decreasing ROS and pro-inflammatory cytokines, and improving QoL [103].
Activation of proteolytic systems
In experimental cancer cachexia the ubitquitin-proteasome-system (UPS) has been shown to play the major role in the degradation of muscular proteins [1, 15, 104, 105]. Increased mRNA levels of ubiquitin and proteasome subunits, as well as increased proteasome activity, have been observed in numerous animal models of cancer cachexia [18, 38, 39, 105–110]. Treatment with proteasome inhibitors was successful in ameliorating cachexia in C26-mice [111]. In animal models the regulation of expression of genes for the E3-ubiquitin ligases, muscle ring finger-1 (MuRF-1) and muscle atrophy F box (MAfxb) has been shown to be increased in different types of muscle atrophy, including cancer cachexia [112–120]. MuRF-1 and MAfxb expression in skeletal muscle are regulated by transcription factors of the FoxO family [38, 62, 112, 113, 121–124] and NF-κB [87, 125]. Furthermore, the activation of the transcription factors Smad2/3 (myostatin pathway) also increases the expression of MuRF-1 and MAfxb via FoxO [4, 58, 59, 61, 62, 104, 113, 121]. Inhibition of MAfxb in fasting mice with muscular atrophy led to decreased expression of myostatin and increased expression of the transcription factor MyoD which is implicated in muscle regeneration [126]. Administration of IL-6 to ApcMin/+ mice induced MAfxb expression via STAT3, whereas MuRF-1 was unaltered at gene and protein levels [35]. These findings suggest that MuRF-1 and MAfxb are involved in different catabolic signaling pathways and represent a common effector.
Compared to these results from animal models, studies of human skeletal muscle have so far produced rather ambivalent results regarding the activation of the UPS in cancer cachexia. Increased activity of the UPS in correlation with disease severity has been demonstrated in skeletal muscle of patients with gastric cancer [34, 127–129], even before the clinical onset of cachexia [128, 129]. Accordingly, it was observed that markers of systemic inflammation (IL-6, CRP) correlated with the increased expression of ubiquitin in skeletal muscle biopsies from cachetic patients with pancreatic cancer, and this increase correlated with the degree of weight loss [130]. Muscle proteolysis and proteasome subunit mRNA were also elevated in colorectal cancer patients compared to controls, whereas after surgical resection of the tumor, the proteolysis rate was reduced to the level of healthy controls [131]. However, a study on patients with pancreatic cancer showed that components of the UPS were only upregulated in subjects with weight loss >10 % [132]. In contrast, studies on lung cancer patients did not find any increased expression of UPS components in skeletal muscle biopsies [133, 134]. Other studies observed that MAfxb expression and MuRF-1 expression were unaltered in muscle biopsies of patients with gastric [135] and colorectal cancer [131]. Interestingly, a study on pancreatic cancer patients with weight loss found that the expression of FoxO1 and −3 was even decreased in skeletal muscle biopsies compared to controls [136].
Similarly, studies investigating the activation of NF-κB in human cancer cachexia have also produced inconsistent results. Whereas the activation of NF-κB has been shown to be an early and sustained event in patients with gastric cancer [137], pre-cachetic lung cancer patients did not show any activation of muscular NF-κB-dependent inflammatory signaling [134]. Furthermore, two recent studies on gene expression profiles in human cancer cachexia showed that none of the previously described genes, including MuRF-1, MAfxb and autophagy related genes (Atgs), were upregulated in skeletal muscle biopsies [138, 139].
Protein-degradation via the authophagy-lysosomal system (ALS) is getting more and more attention in the context of cancer cachexia recently, and its regulation has been shown to overlap with the UPS. Under physiological conditions, the ALS contributes to cell survival and adaption to stress through the controlled degradation of dysfunctional intracellular organelles and proteins by lysosomal proteases (cathepsins) [140]. The regulation of autophagy induction and autophagosome formation is dependent on gene expression of Atgs. In animal models, Atgs (e.g. Atg7, LC3B, Bnip3, Beclin-1) have been shown to be over-expressed during muscle atrophy [140–143] and are partly regulated by FoxO3 [121, 143, 144] and p38-MAPK [145, 146]. Furthermore, elevated levels of total cathepsin activity have been observed in hepatoma-bearing rats and treatment with a cathepsin-inhibitor was able to attenuate muscle depletion [147, 148]. A recent study by Penna et al. demonstrated that autophagy is increased in C26-bearing mice as well as in Yoshida-hepatoma rats and LLC mice [149].
However, in skeletal muscle of patients with early stages of lung cancer, mRNA levels of the lysosomal proteases cathepsin B and D were elevated, whereas components of the UPS were not increased [133]. Furthermore, cathepsin B mRNA levels correlated with fat-free mass index and tumor stage and were higher in cancer patients who were smokers. Since these observations were made before the clinical onset of cachexia, it is suggested that similar to findings from animal models, cathepsin B expression is involved in the induction of cachexia in lung cancer patients [133]. Increased levels of cathepsin D have been observed in cancer patients with other tumor entities as well [150].
Decreased protein synthesis
In addition to the increased protein degradation, it has been postulated that muscle atrophy in cancer cachexia is also due to decreased protein synthesis [78, 141]. Under physiological conditions, activation of the anabolic PI3K/Akt/mTOR pathway results in down-regulation of MuRF-1 and MAfxb through inhibition of FoxO [12, 119, 136] and simultaneous stimulation of protein synthesis by activation of mammalian target of rapamycin (mTOR) and glycogen synthase kinase 3β (GSK3β) [12, 119, 136, 151]. The PI3K/Akt/mTOR signaling cascade is activated by insulin or IGF-1. Low levels of insulin or IGF-1 and elevated levels of glucocorticoids induce the loss of muscle protein in diabetes, and insulin resistance is a characteristic feature of many systemic diseases with muscle wasting [152].
Activation of the protein kinase Akt was shown to be decreased in muscle and adipose tissue of tumor-bearing, cachectic mice [124]. In contrast, a recent study on cancer cachexia in mice observed an increase in Akt activation, while mTOR signaling and FoxO activity were suppressed [38]. However, Penna et al. observed no decreased activity of Akt in two distinct animal models of cancer cachexia [42]. Downstream of Akt, no suppression of protein synthesis was observed, with levels of activated p70S6K and GSK3-β being normal or increased, and levels of eIF2α being decreased [42]. Other studies even found that mTOR signaling was activated rather than suppressed during cancer cachexia. One hypothesis is that mTOR may be activated through the intracellular concentration of free amino acids, which rises when protein degradation is increased [153]. According to another hypothesis a certain increase in protein synthesis is required for protein degradation, and the PI3K/Akt/mTOR pathway has a dual role. When mTOR was inhibited 30 min before the application of PIF, protein degradation was not increased, suggesting that increased proteolysis requires the activation of mTOR [85]. In addition, Robert et al. showed that beyond stimulating the synthesis of muscle protein, mTOR also regulated the production of pro-cachetic factors such as IL-6 and IL-10, and inhibition of mTOR with rapamycin resulted in reduced IL-10 mRNA translation and ameliorated the cachectic phenotype [154].
Available data on the role and activation status of the PI3K/Akt/mTOR pathway in cancer patients is very limited. In one study on patients with pancreatic cancer and weight loss, decreased protein levels and activation of Akt, mTOR, p70S6K, GSK3-ß and FoxO1 were observed in skeletal muscle tissue compared to samples from patients with pancreatic cancer without weight loss [136]. Another study on patients with GIT-cancer and weight loss found increased protein levels and phosphorylation of PKR and eIF2α. Myosin levels decreased as the weight loss increased. The linear relationship between myosin expression and the extent of phosphorylation of eIF2α and PKR suggests that the phosphorylation of PKR may be an important initiator of muscle wasting in cancer patients [155]. In addition, a study on patients with colorectal cancer investigated the pattern of muscle protein turnover before and after surgical tumor resection compared to healthy controls and evaluated the anabolic response of skeletal muscle tissue to nutrition in three groups (control, pre-operative, post-operative). The authors observed that myofibrillar protein synthesis increased after feeding of healthy controls, whereas there was no response in the preoperative cancer patient group. After surgery the anabolic response to feeding was recovered. However, in the healthy control group and in the preoperative patient group, nutrition led to a significant increase in p70S6K and 4E-BP1 phosphorylation, whereas in the postoperative patient group nutrition did not lead to this effect. The phosphorylation of Akt was unchanged in all groups. Furthermore, increased expression of proteasome subunit mRNA was observed in the preoperative group compared to controls, but interestingly MuRF-1 and MAfxb expression were unchanged in all groups [131].
Inhibition of muscle regeneration
Finally, the inhibition of positive regulators of muscle growth and regeneration factors (MRFs) also plays an important role in cancer cachexia. The transcription factor MyoD is an essential regulator of myogenesis and myoblast differentiation, and is crucial for regeneration of muscle tissue from satellite cells [14, 156, 157]. MyoD was shown to be inhibited by pro-inflammatory cytokines, myostatin and PIF. The proteolysis of MyoD during muscle atrophy was shown to be effectuated through ubiquitination by MAfxb in vitro and in vivo, and atrophy was attenuated by inhibition of this process [156].
Data on the impairment of MRFs in human cachexia patients is very limited. A study on the expression of genes involved in muscle regeneration (Pax7, MyoD, Myf5, nMyHC, necdin) in gastric cancer patients found that Pax7 was significantly increased in all disease stages compared to controls, whereas MyoD and necdin were only upregulated in early disease stages [10]. Pax7 was also shown to be dysregulated in muscle biopsies of patients with pancreatic cancer [158]. These results suggest that catabolic signals possibly stimulate a counteractive regenerative response in satellite cells. However, it is suggested that the regenerative response is dysfunctional during cancer-induced muscle wasting. Furthermore, these results show a possible protective role of the protein necdin, which has previously been shown to prevent cachexia in a mouse model [10].
Discussion: signaling pathways leading to skeletal muscle mass in cancer cachexia – can findings from animal models be translated to humans?
General issues in cancer cachexia research and clinical trials
The results presented in this review underline the fact that human cancer cachexia is a heterogeneous clinical syndrome with many variable contributing factors, all of which cannot be reflected by an animal model. Advantages of animal models clearly are the homogeneity of study subjects and the possibility to effectively control influencing confounders, e.g. by accurate control of diet or exercise and the use of pair fed controls [159]. However, none of the numerous animal models is ideal to simulate the complex biology behind human cancer cachexia. Too many variables are playing a role, including tumor biology and location, host-tumor interactions, co-morbidities, prior anti-cancer therapies and psychosocial issues. For example, implanted tumors in syngeneic animal models are well defined and do not metastasize, which does not accurately reflect the growth of malignant human tumors. Furthermore, the tumors are usually implanted in very young animals, which undergo rapid tumor growth up to a size of more than 10 % of the body mass and severe wasting within days to weeks. In contrast, growth and wasting are less aggressive in humans, where cachexia usually occurs within months to years and tumors usually don’t grow to more than 1 % of body mass [35, 160]. Xenograft models resemble more to human tumors, but lack of the interaction between tumor and immune system due to the needed immunosuppression of the animals. Genetically engineered animals develop tumors spontaneously and reproduce naïve tumor-host interactions, but the genetic alterations are expressed in all tissue, which is also not the case in human tumorigenesis. Carcinogen-induced tumors in animals probably reflect normal tumor growth and tumor-host interactions most closely, but are tedious and costly [54]. However, since none of the models addresses all aspects of human cancer cachexia, use of a combination is recommended, and careful consideration of these issues is needed before translation to clinical research can be made [54].
Compared to the paucity of literature on experimental cancer cachexia, studies on human samples are relatively scarce. This might be an accessibility issue, which is why the cooperation of researchers with surgical departments who can easily provide intraoperative muscle biopsies from cancer patients, should be encouraged.
Another general issue in cachexia research is the difficulty of recruiting a homogeneous study cohort of cancer patients. The clinical presentation of cachexia is becoming more and more variable in the context of the growing proportion of obese patients [12, 161]. Furthermore, skeletal muscle mass is generally greater in men than in women, but paradoxically loss of muscle and lean body mass is also greater in men than in women, which is possibly due to hormonal differences [12, 162]. There is also a great heterogeneity in energy expenditure between patients. As observed by Knox et al., cancer patients can be hypermetabolic, normal or hypometabolic [12, 160, 163]. Decreased physical activity combined with increased REE is another very individual and confounding element. Cancer patients are often prone to physical inactivity due to their age and co-morbidities. It is known that bed rest by itself causes muscle wasting by amplifying catabolism and desensitizing muscle cells for anabolic signals [161]. In addition, anorexia, reduced food intake, psychosocial issues and aggressive anti-cancer therapies contribute to weight loss and cachexia in human cancer patients [164]. Several modern anti-cancer therapies target signaling pathways which are important for tumorigenesis, such as Akt/mTOR, but also regulate protein anabolism in skeletal muscle and other tissues, so muscle wasting can probably be partially attributed to chemotherapy [161]. Finally, it has to be assumed that there is a genetic contribution to cachexia as well, since even with the same tumor type and stage, some individuals develop cachexia whereas other do not [12]. Studies trying to identify genes for a predisposition to cancer cachexia have shown that single nucleotide polymorphisms in cytokine genes have been associated with the prevalence of cachexia [12]. In addition, polymorphisms in the vitamin D receptor have been proposed as early clinical predictors of the development of a more aggressive form of cachexia in cancer patients [165]. Furthermore, the 1082G allele in the IL-10 promoter [166] and the C allele of the rs6136 polymorphism in the P-selectin gene have been validated as pro-cachectic genotypes [167]. These findings point towards the close relationship between the innate immune system and cancer cachexia. However, genome-wide studies are currently lacking, and the role of genetic predisposition has not yet been fully clarified [168].
Further problems are encountered in the design of clinical trials for cancer cachexia therapies. How to approach such studies has been extensively discussed as a result of the fact that many randomized controlled trials were conducted without resulting in any approved therapies [164]. A general consensus over the definition and use of diagnostic criteria of cancer cachexia and its different stages has still not been reached. The use of new, much more specific and sensitive measuring techniques of body composition, using computed tomography (CT) imaging and novel tracer techniques with labeled amino acids, will allow precise quantification of muscle protein kinetics and will facilitate the definition of more specific endpoints and guide the evaluation of pharmaceutical interventions [169]. A recent study comparing different diagnostic and assessment criteria for cachexia in a cohort of patients with advanced colorectal cancer found that the Cancer Cachexia Study Group’s cachexia score was the best prognostic factor for overall survival [170]. This score includes three diagnostic criteria: (1) weight loss >10 %, (2) intake <1500 kcal/d, and (3) CRP >10 mg/L. However, it does not include assessment of skeletal muscle mass, which is increasingly seen as the primary indicator of cachexia in the current literature. More and more studies use CT images for the quantification and observation of muscle wasting in cancer cachexia and have shown a specific association between muscle loss and reduced survival [161, 171]. To conduct comparable clinical trials in the future, a clear definition of criteria for cachexia is indispensable.
Translation of findings from animal models to human cancer cachexia therapy
Even though animal models are not sufficient to mimic all complex aspects of cachexia in cancer patients, results from these pre-clinical studies have already led to a substantial number of potential therapeutic targets and approaches.
Anti-cytokine treatments
In most animal studies, serum elevation of pro-inflammatory cytokines like TNF-α and IL-6 is associated with muscle wasting and anti-cytokine treatments showed great promise. Unfortunately, in humans the results from investigations of the role of cytokines are completely inconsistent. Most clinical trials of inhibitors of synthesis or activity of TNF-α have so far not proven to be effective in preserving lean body mass in cancer patients [9, 14, 20]. Thalidomide, an inhibitor of TNF-α and other pro-inflammatory cytokines, was shown to be effective in the treatment of cancer cachexia in patients with GIT cancers but has strong adverse side effects [161, 172–174]. Anti-TNF-α antibodies such as infliximab and etanercept did not show any significant improvements in cachectic patients and were not well tolerated either [175, 176].
Preclinical and clinical (phase I and II) studies performed on the IL-6 antibody ALD518 in patients with non-small cell lung cancer (NSCLC) showed that this treatment has the potential to improve anemia, reduce cancer-related cachexia and ameliorate fatigue, while having minimal adverse effects [173, 177]. Other anti-IL-6 antibodies or inhibitors (BMS 945429, selumetinib) have also been shown to be well tolerated and improve fatigue and loss of lean body mass [177, 178].
Currently, no anti-cytokine therapies are approved for the treatment of cancer cachexia and further clinical trials are needed to confirm their benefits. However, some clinical studies have shown that anti-cytokine treatments have the potential to ameliorate combination therapy protocols [9, 12].
The discrepancies between animal and human studies could be partly due to differences and difficulties in measuring serum cytokine levels or simply reflect the heterogeneity of the individual cytokine response in different types of cancers and patients [16]. Furthermore, pro-inflammatory mediators (especially IL-1) not only take effect in the inflammatory reaction but also have central effects leading to reduced food intake and anorexia in a complex and individual interaction with various hormones and neuropeptides. Another recently identified source of these discrepancies could be that different C26 tumor cell lines secret different amounts of IL-6 depending on sample storage conditions and the number of cell passages in vitro [179]. These results were reproduced in vivo and could implicate that measurements of IL-6 secretion in human cancer cachexia samples from different laboratories might largely vary depending on the treatment and storage conditions of samples.
Myostastin/ActRIIB targeting treatments
Blocking of myostatin and ActRIIB signaling showed very promising results in animal and in vitro studies. Several clinical approaches are currently being evaluated by pharmaceutical companies, but results are still lacking [180]. In cachexia due to heart failure, myostatin expression has been shown to be upregulated in animals [181] and patients [182]. In addition, increased myostatin expression has repeatedly been seen in cachectic patients secondary to HIV/AIDS or severe COPD [60]. Furthermore, mice with a heart-specific knockout of the myostatin gene appear to be resistant to skeletal muscle loss, which indicates that targeting this pathway could be of benefit for patients with muscle wasting in the context of chronic heart failure and maybe also in cancer cachexia [183, 184]. ActRIIB receptor and myostatin inhibitors are currently being evaluated in pre-clinical trials of muscle wasting and degenerative disorders. Among the first agents developed for clinical settings are the monoclonal anti-myostatin antibodies, which are currently undergoing phase II trials in patients with NSCLC and pancreatic cancer [161].
PIF/AngII targeting treatments
Proteolyis inducing factor (PIF) and AngII are important cachectic factors in experimental cancer wasting as well. In humans, however, the existence of a homologue to the murine PIF is still debated. Wieland et al. questioned the existence of human PIF after finding neither a correlation between the presence of this protein in urine and weight loss or survival of cancer patients, nor a specificity for malignant diseases, since they found the same protein in patients with cachexia due to congestive heart failure [89]. However, their study was later criticized for its methodology, since a cross-reactive antibody was used and western blot bands were not correctly confirmed [185]. In the end, results point towards a role of PIF, especially in GIT cancer, but its relevance still remains to be confirmed, and the source and mode of action of this protein need to be determined. Concerning the role of AngII in the pathophysiology of cancer cachexia, there are currently no published studies on human material. However, treatment with ACE inhibitors was found to ameliorate cardiac cachexia, and there are reports of unpublished results from clinical trials started by pharmaceutical companies which point to a possible benefit of this treatment in cancer cachexia [102]. This hypothesis should be confirmed in randomized controlled trials.
Treatments targeting increased proteolysis and decreased protein synthesis
Components of proteolytic systems were activated in most experimental models of cancer cachexia. However, in cancer patients these observations were not consistently confirmed, suggesting that different types of tumors and individual hosts may produce different reactions of the proteolytic systems. However, current attempts to pharmaceutically block the enhanced activity of the UPS by using antagonists of the inducers of proteasome expression, inhibitors of NF-κB signaling and inhibitors of ubiquitin ligases or proteasome subunits have not yet yielded any approved treatment options [99, 186]. Inhibitors of NF-κB, such as resveratrol, thalidomide, ibuprofen, eicosapentaenoic acid, and beta-hydroxy-beta-methylbutyrate, have been shown to improve skeletal muscle mass in cachexia. In addition, proteasome inhibitors have been shown to have positive effects in Duchenne and Becker muscular dystrophy. However, in a study on patients with pancreatic cancer, treatment with bortezomib showed no beneficial effects on cachexia [187].
There is still much debate over whether the predominant component of muscle loss during cachexia is increased proteolysis or decreased protein synthesis. However, the anabolic Akt/mTOR pathway was identified as the most important anabolic cascade, and cross-talk with proteolytic pathways was demonstrated in animal models. However, some studies found protein synthesis to be activated rather than suppressed. This might reflect the fact that cachexia is a dynamic process, passing through different stages that might be dominated either by protein breakdown or protein synthesis. For example, it is possible that during certain stages of cachexia, protein synthesis is actually increased in skeletal muscle due to the local production of cytokines or as a counter-regulatory phenomenon. In this context, it becomes clear why it is hard to interpret human studies on cancer cachexia, which usually include heterogeneous subjects in terms of the stage of cachexia. To overcome this problem it is essential that future studies accurately define different stages of cachexia so their results can be stratified accordingly.
The impairment of muscle regeneration is a relatively new aspect of muscle loss in cancer cachexia and, as presented above, data on its role in human cancer cachexia remains very limited. To our knowledge no drug targets have been preselected in this field and further research is needed to identify and test those.
Current cancer cachexia therapy options
Considering the multidimensional background of cancer cachexia, it is more and more the accepted view that multimodal therapeutic approaches, including exercise, nutrient supplementation, appetite stimulation and pharmacological intervention, have to be implemented and individually tailored for patients at different stages of cachexia [5, 161]. A large-scale meta-analysis showed that nutritional interventions were successful in increasing energy intake, body weight and some aspects of QoL [188]. The evidence for interventions with resistance exercise training is not as extensive yet, but first results are promising [189]. Nutrient supplementation with N3-fatty acids, e.g. eicosapentaenoic acid or fish oil, also have shown positive effects on muscle loss and survival; however, the evidence is not yet sufficient for recommendation [161]. In addition, improving patients’ metabolism by insulin or metformin treatment was shown to increase whole body fat (without counteracting muscle loss) and survival in initial study results [161, 190]. Moreover, secondary symptoms like pain, diarrhea or stomatitis have to be managed correctly to evaluate the efficacy of new treatments of cancer cachexia [161, 164]. Additional multidimensional pharmacological therapy should ideally include drugs that target the inflammatory status, oxidative stress, nutritional disorders, muscle catabolism, anemia, immunosuppression, and fatigue [173]. Anti-inflammatory drugs like COX inhibitors (indomethacin, ibuprofen) not only reduce the inflammatory response but also have a positive effect on REE and were shown to prolong survival in malnourished patients with advanced cancer [191]. Finally, careful psychosocial counseling and access to self-help groups should be provided [173]. Moreover, successful surgical removal of the tumor and/or oncological treatments should be the starting point for rehabilitation of patients with cancer-associated muscle wasting [160].
Conclusion
In conclusion, given the heterogeneous and multi-factorial etiology of cachexia, it is likely that this syndrome is a result of deregulation of multiple signaling pathways. It is possible that certain pathways are involved only in a subset of patients and to an individual extent, which would determine if the patient responds to therapeutic interventions on the level of intracellular signaling pathways. However, there is also a belief that treatments that preserve muscle mass per se can improve survival and QoL of cancer patients, regardless of the underlying molecular mechanisms. This review shows that, even though animal models cannot imitate all of the complex aspects of human cancer cachexia, they provide a robust setting to develop and test new, targeted therapies. Ultimately, further studies on the signaling pathways that lead to skeletal muscle loss in larger and more homogeneous cohorts of human patients are warranted to confirm potential drug targets identified in experimental animal models.
Abbreviations
- GIT:
-
gastrointestinal tract
- COPD:
-
chronic obstructive pulmonary disease
- QoL:
-
quality of life
- BMI:
-
body mass index
- CRP:
-
C-reactive protein
- REE:
-
resting energy expenditure
- ATP:
-
adenosin-triphosphate
- UCP:
-
uncoupling protein
- TNF-α:
-
tumor necrosis factor-alpha
- LLC:
-
Lewis lung cell carcinom
- TRAF-6:
-
TNF-receptor adaptor protein
- TNFR:
-
TNF-receptor
- mRNA:
-
messenger ribonucleic acid
- NF-κB:
-
nuclear factor κB
- IL:
-
interleukin
- NSCLC:
-
non-small cell lung cancer
- INF-γ:
-
interferon gamma
- VEGF:
-
vascular endothelial growth factor
- TGF:
-
transforming growth factor
- GDF:
-
growth differentiation factor
- ActRIIB:
-
activin type-2 receptor
- IGF:
-
insulin-like growth factor
- MIC:
-
macrophage inhibitory cytokine
- PIF:
-
proteolysis-inducing factor
- UPS:
-
ubiquitin proteasome system
- PKR:
-
RNA-dependent protein kinase
- HCAP:
-
human cachexia-associated protein
- Ang II:
-
angiotensin-II
- REE:
-
resting energy expenditure
- ROS:
-
reactive oxygen species
- ACE:
-
angiotensin-converting enzyme
- MuRF-1:
-
muscle ring finger-1
- MAfxb:
-
muscle atrophy F box
- Atgs:
-
autophagy related genes
- ALS:
-
authophagy-lysosomal system
- GSK:
-
glycogen synthase kinase
- mTOR:
-
mammalian target of rapamycin
- MRF:
-
muscle growth and regeneration factor
- CT:
-
computed tomography
- HIV/AIDS:
-
human immunodeficiency virus/ acquired immune deficiency syndrome
- COX:
-
cyclooxygenase
References
Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89(2):381–410.
Fearon KC. Cancer cachexia: developing multimodal therapy for a multidimensional problem. Eur J Cancer. 2008;44(8):1124–32.
Laviano A, Meguid MM, Inui A, Muscaritoli M, Rossi-Fanelli F. Therapy insight: Cancer anorexia-cachexia syndrome--when all you can eat is yourself. Nat Clin Pract Oncol. 2005;2(3):158–65.
Tisdale MJ. Reversing cachexia. Cell. 2010;142(4):511–2.
Tsoli M, Robertson G: Cancer cachexia: malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol Metab. 2013;24(4):174-83. doi: 10.1016/j.tem.2012.10.006. Epub 29 Nov 2012.
Donohoe CL, Ryan AM, Reynolds JV. Cancer cachexia: mechanisms and clinical implications. Gastroenterol Res Pract. 2011;2011:601434.
Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333(6039):233–8.
Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12(5):489–95.
Penna F, Minero VG, Costamagna D, Bonelli G, Baccino FM, Costelli P. Anti-cytokine strategies for the treatment of cancer-related anorexia and cachexia. Expert Opin Biol Ther. 2010;10(8):1241–50.
Pessina P, Conti V, Pacelli F, Rosa F, Doglietto GB, Brunelli S, et al. Skeletal muscle of gastric cancer patients expresses genes involved in muscle regeneration. Oncol Rep. 2010;24(3):741–5.
Stephens NA, Gray C, MacDonald AJ, Tan BH, Gallagher IJ, Skipworth RJ, et al. Sexual dimorphism modulates the impact of cancer cachexia on lower limb muscle mass and function. Clin Nutr. 2012;31(4):499–505.
Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 2012;16(2):153–66.
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142(4):531–43.
Bossola M, Pacelli F, Tortorelli A, Doglietto GB. Cancer cachexia: it’s time for more clinical trials. Ann Surg Oncol. 2007;14(2):276–85.
Fanzani A, Conraads VM, Penna F, Martinet W. Molecular and cellular mechanisms of skeletal muscle atrophy: an update. J Cachex Sarcopenia Muscle. 2012;3(3):163–79.
Patra SK, Arora S. Integrative role of neuropeptides and cytokines in cancer anorexia-cachexia syndrome. Clini Chim Acta. 2012;413(13–14):1025–34.
Catalano MG, Fortunati N, Arena K, Costelli P, Aragno M, Danni O, et al. Selective up-regulation of tumor necrosis factor receptor I in tumor-bearing rats with cancer-related cachexia. Int J Oncol. 2003;23(2):429–36.
Llovera M, Garcia-Martinez C, Lopez-Soriano J, Agell N, Lopez-Soriano FJ, Garcia I, et al. Protein turnover in skeletal muscle of tumour-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett. 1998;130(1–2):19–27.
Costelli P, Bossola M, Muscaritoli M, Grieco G, Bonelli G, Bellantone R, et al. Anticytokine treatment prevents the increase in the activity of ATP-ubiquitin- and Ca(2+)-dependent proteolytic systems in the muscle of tumour-bearing rats. Cytokine. 2002;19(1):1–5.
Hitt A, Graves E, McCarthy DO. Indomethacin preserves muscle mass and reduces levels of E3 ligases and TNF receptor type 1 in the gastrocnemius muscle of tumor-bearing mice. Res Nurs Health. 2005;28(1):56–66.
Zhou W, Jiang ZW, Tian J, Jiang J, Li N, Li JS. Role of NF-kappaB and cytokine in experimental cancer cachexia. World J Gastroenterol. 2003;9(7):1567–70.
Baracos VE. Regulation of skeletal-muscle–protein turnover in cancer-associated cachexia. Nutrition. 2000;16(10):1015–8.
Paul PK, Kumar A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy. 2011;7(5):555–6.
Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, et al. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol. 2012;32(7):1248–59.
Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191(7):1395–411.
Kumar A, Bhatnagar S, Paul PK. TWEAK and TRAF6 regulate skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2012;15(3):233–9.
Karayiannakis AJ, Syrigos KN, Polychronidis A, Pitiakoudis M, Bounovas A, Simopoulos K. Serum levels of tumor necrosis factor-alpha and nutritional status in pancreatic cancer patients. Anticancer Res. 2001;21(2B):1355–8.
Nakashima J, Tachibana M, Ueno M, Miyajima A, Baba S, Murai M. Association between tumor necrosis factor in serum and cachexia in patients with prostate cancer. Clin Cancer Res. 1998;4(7):1743–8.
Pfitzenmaier J, Vessella R, Higano CS, Noteboom JL, Wallace Jr D, Corey E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer. 2003;97(5):1211–6.
Wang YY, Lo GH, Lai KH, Cheng JS, Lin CK, Hsu PI. Increased serum concentrations of tumor necrosis factor-alpha are associated with disease progression and malnutrition in hepatocellular carcinoma. J Chin Med Assoc. 2003;66(10):593–8.
Ariapart P, Bergstedt-Lindqvist S, van Harmelen V, Permert J, Wang F, Lundkvist I. Resection of pancreatic cancer normalizes the preoperative increase of tumor necrosis factor alpha gene expression. Pancreatology. 2002;2(5):491–4.
Kemik O, Sumer A, Kemik AS, Hasirci I, Purisa S, Dulger AC, et al. The relationship among acute-phase response proteins, cytokines and hormones in cachectic patients with colon cancer. World J Surg Oncol. 2010;8:85.
Kemik O, Kemik AS, Begenik H, Erdur FM, Emre H, Sumer A, et al. The relationship among acute-phase responce proteins, cytokines, and hormones in various gastrointestinal cancer types patients with cachectic. Hum Exp Toxicol. 2012;31(2):117–25.
Sun YS, Ye ZY, Qian ZY, Xu XD, Hu JF. Expression of TRAF6 and ubiquitin mrna in skeletal muscle of gastric cancer patients. J Exp Clin Cancer Res. 2012;31(1):81.
Carson JA, Baltgalvis KA. Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev. 2010;38(4):168–76.
Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One. 2011;6(7):e22538.
Baltgalvis KA, Berger FG, Pena MM, Davis JM, White JP, Carson JA. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc (Min/+) mouse. Pflugers Arch - Eur J Physiol. 2009;457(5):989–1001.
White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One. 2011;6(9):e24650.
Fujita J, Tsujinaka T, Yano M, Ebisui C, Saito H, Katsume A, et al. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int J Cancer. 1996;68(5):637–43.
Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab. 2012;303(3):E410–21.
Alvarez B, Quinn LS, Busquets S, Quiles MT, Lopez-Soriano FJ, Argiles JM. Tumor necrosis factor-alpha exerts interleukin-6-dependent and -independent effects on cultured skeletal muscle cells. Biochim Biophys Acta. 2002;1542(1–3):66–72.
Penna F, Bonetto A, Muscaritoli M, Costamagna D, Minero VG, Bonelli G, et al. Muscle atrophy in experimental cancer cachexia: is the IGF-1 signaling pathway involved? Int J Cancer. 2010;127(7):1706–17.
Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br J Cancer. 1996;73(12):1560–2.
Scheede-Bergdahl C, Watt HL, Trutschnigg B, Kilgour RD, Haggarty A, Lucar E, et al. Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin Nutr. 2012;31(1):85–8.
Yeh KY, Li YY, Hsieh LL, Lu CH, Chou WC, Liaw CC, et al. Analysis of the effect of serum interleukin-6 (IL-6) and soluble IL-6 receptor levels on survival of patients with colorectal cancer. Jpn J Clin Oncol. 2010;40(6):580–7.
Krzystek-Korpacka M, Matusiewicz M, Diakowska D, Grabowski K, Blachut K, Kustrzeba-Wojcicka I, et al. Impact of weight loss on circulating IL-1, IL-6, IL-8, TNF-alpha, VEGF-A, VEGF-C and midkine in gastroesophageal cancer patients. Clin Biochem. 2007;40(18):1353–60.
Kim HJ, Kim HJ, Yun J, Kim KH, Kim SH, Lee SC, et al. Pathophysiological role of hormones and cytokines in cancer cachexia. J Korean Med Sci. 2012;27(2):128–34.
Ebrahimi B, Tucker SL, Li D, Abbruzzese JL, Kurzrock R. Cytokines in pancreatic carcinoma: correlation with phenotypic characteristics and prognosis. Cancer. 2004;101(12):2727–36.
Kuroda K, Nakashima J, Kanao K, Kikuchi E, Miyajima A, Horiguchi Y, et al. Interleukin 6 is associated with cachexia in patients with prostate cancer. Urology. 2007;69(1):113–7.
Martin F, Santolaria F, Batista N, Milena A, Gonzalez-Reimers E, Brito MJ, et al. Cytokine levels (IL-6 and IFN-gamma), acute phase response and nutritional status as prognostic factors in lung cancer. Cytokine. 1999;11(1):80–6.
Iwase S, Murakami T, Saito Y, Nakagawa K. Steep elevation of blood interleukin-6 (IL-6) associated only with late stages of cachexia in cancer patients. Eur Cytokine Netw. 2004;15(4):312–6.
Martignoni ME, Kunze P, Hildebrandt W, Kunzli B, Berberat P, Giese T, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin Cancer Res. 2005;11(16):5802–8.
Wigmore SJ, Fearon KC, Sangster K, Maingay JP, Garden OJ, Ross JA. Cytokine regulation of constitutive production of interleukin-8 and −6 by human pancreatic cancer cell lines and serum cytokine concentrations in patients with pancreatic cancer. Int J Oncol. 2002;21(4):881–6.
Bennani-Baiti N, Walsh D. Animal models of the cancer anorexia-cachexia syndrome. Support Care Cancer. 2011;19(9):1451–63.
Maltoni M, Fabbri L, Nanni O, Scarpi E, Pezzi L, Flamini E, et al. Serum levels of tumour necrosis factor alpha and other cytokines do not correlate with weight loss and anorexia in cancer patients. Support Care Cancer. 1997;5(2):130–5.
Jatoi A, Egner J, Loprinzi CL, Sloan JA, Novotny PJ, Dakhil SR, et al. Investigating the utility of serum cytokine measurements in a multi-institutional cancer anorexia/weight loss trial. Support Care Cancer. 2004;12(9):640–4.
Dulger H, Alici S, Sekeroglu MR, Erkog R, Ozbek H, Noyan T, et al. Serum levels of leptin and proinflammatory cytokines in patients with gastrointestinal cancer. Int J Clin Pract. 2004;58(6):545–9.
Han HQ, Mitch WE. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr Opin Support Palliat Care. 2011;5(4):334–41.
Argiles JM, Orpi M, Busquets S, Lopez-Soriano FJ. Myostatin: more than just a regulator of muscle mass. Drug Discov Today. 2012;17(13–14):702–9.
Elliott B, Renshaw D, Getting S, Mackenzie R. The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol. 2012;205(3):324–40.
Lokireddy S, Wijesoma IW, Bonala S, Wei M, Sze SK, McFarlane C, et al. Myostatin is a novel tumoral factor that induces cancer cachexia. Biochem J. 2012;446(1):23–36.
Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13(3):225–9.
Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296(6):C1258–70.
Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297(5):C1124–32.
Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachex Sarcopenia Muscle. 2011;2(3):143–51.
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831–40.
Murphy KT, Chee A, Gleeson BG, Naim T, Swiderski K, Koopman R, et al. Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol Regul Integr Comp Physiol. 2011;301(3):R716–26.
Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296(6):C1248–57.
Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun. 2010;391(3):1548–54.
Mathew SJ. InACTIVatINg cancer cachexia. Dis Model Mech. 2011;4(3):283–5.
Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of Cachexia in Mice by Systemically Administered Myostatin. Science. 2002;296(5572):1486–8.
Trendelenburg AU, Meyer A, Jacobi C, Feige JN, Glass DJ. TAK-1/p38/nNFkappaB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skelet Muscle. 2012;2(1):3.
Li L, Shen JJ, Bournat JC, Huang L, Chattopadhyay A, Li Z, et al. Activin signaling: effects on body composition and mitochondrial energy metabolism. Endocrinology. 2009;150(8):3521–9.
Gold E, Marino FE, Harrison C, Makanji Y, Risbridger G: Activin-betac reduces reproductive tumour progression and abolishes cancer-associated cachexia in inhibin deficient mice. J Pathol. 2013;229(4):599-607. doi: 10.1002/path.4142. Epub 25 Jan 2013.
Li Q, Kumar R, Underwood K, O’Connor AE, Loveland KL, Seehra JS, et al. Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Mol Hum Reprod. 2007;13(9):675–83.
Tsai VW, Husaini Y, Manandhar R, Lee-Ng KK, Zhang HP, Harriott K, et al. Anorexia/cachexia of chronic diseases: a role for the TGF-beta family cytokine MIC-1/GDF15. J Cachex Sarcopenia Muscle. 2012;3(4):239–43.
Aversa Z, Bonetto A, Penna F, Costelli P, Di Rienzo G, Lacitignola A, et al. Changes in myostatin signaling in non-weight-losing cancer patients. Ann Surg Oncol. 2012;19(4):1350–6.
Eley HL, Tisdale MJ. Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J Biol Chem. 2007;282(10):7087–97.
Mirza KA, Wyke SM, Tisdale MJ. Attenuation of muscle atrophy by an N-terminal peptide of the receptor for proteolysis-inducing factor (PIF). Br J Cancer. 2011;105(1):83–8.
Russell ST, Eley H, Tisdale MJ. Role of reactive oxygen species in protein degradation in murine myotubes induced by proteolysis-inducing factor and angiotensin II. Cell Signal. 2007;19(8):1797–806.
Lorite MJ, Smith HJ, Arnold JA, Morris A, Thompson MG, Tisdale MJ. Activation of ATP-ubiquitin-dependent proteolysis in skeletal muscle in vivo and murine myoblasts in vitro by a proteolysis-inducing factor (PIF). Br J Cancer. 2001;85(2):297–302.
Wyke SM, Tisdale MJ. NF-kappaB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin-proteasome system in skeletal muscle. Br J Cancer. 2005;92(4):711–21.
Whitehouse AS, Tisdale MJ. Increased expression of the ubiquitin-proteasome pathway in murine myotubes by proteolysis-inducing factor (PIF) is associated with activation of the transcription factor NF-kappaB. Br J Cancer. 2003;89(6):1116–22.
Argiles JM, Busquets S, Garcia-Martinez C, Lopez-Soriano FJ. Mediators involved in the cancer anorexia-cachexia syndrome: past, present, and future. Nutrition. 2005;21(9):977–85.
Russell ST, Eley HL, Wyke SM, Tisdale MJ. Involvement of phosphoinositide 3-kinase and Akt in the induction of muscle protein degradation by proteolysis-inducing factor. Biochem J. 2008;409(3):751–9.
Tisdale MJ. Catabolic mediators of cancer cachexia. Curr Opin Support Palliat Care. 2008;2(4):256–61.
Cai D, Frantz JD, Tawa Jr NE, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119(2):285–98.
Eley HL, Russell ST, Tisdale MJ. Attenuation of muscle atrophy in a murine model of cachexia by inhibition of the dsRNA-dependent protein kinase. Br J Cancer. 2007;96(8):1216–22.
Wieland BM, Stewart GD, Skipworth RJ, Sangster K, Fearon KC, Ross JA, et al. Is there a human homologue to the murine proteolysis-inducing factor? Clin Cancer Res. 2007;13(17):4984–92.
Wigmore SJ, Todorov PT, Barber MD, Ross JA, Tisdale MJ, Fearon KC. Characteristics of patients with pancreatic cancer expressing a novel cancer cachectic factor. Br J Surg. 2000;87(1):53–8.
Cabal-Manzano R, Bhargava P, Torres-Duarte A, Marshall J, Bhargava P, Wainer IW. Proteolysis-inducing factor is expressed in tumours of patients with gastrointestinal cancers and correlates with weight loss. Br J Cancer. 2001;84(12):1599–601.
Williams ML, Torres-Duarte A, Brant LJ, Bhargava P, Marshall J, Wainer IW. The relationship between a urinary cachectic factor and weight loss in advanced cancer patients. Cancer Investig. 2004;22(6):866–70.
Wang Z, Corey E, Hass GM, Higano CS, True LD, Wallace Jr D, et al. Expression of the human cachexia-associated protein (HCAP) in prostate cancer and in a prostate cancer animal model of cachexia. Int J Cancer. 2003;105(1):123–9.
Jatoi A, Foster N, Wieland B, Murphy B, Nikcevich D, LaPlant B, et al. The proteolysis-inducing factor: in search of its clinical relevance in patients with metastatic gastric/esophageal cancer. Dis Esophagus. 2006;19(4):241–7.
Sanders PM, Russell ST, Tisdale MJ. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br J Cancer. 2005;93(4):425–34.
Song YH, Li Y, Du J, Mitch WE, Rosenthal N, Delafontaine P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest. 2005;115(2):451–8.
Yoshida T, Semprun-Prieto L, Sukhanov S, Delafontaine P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol. 2010;298(5):H1565–70.
Russell ST, Wyke SM, Tisdale MJ. Mechanism of induction of muscle protein degradation by angiotensin II. Cell Signal. 2006;18(7):1087–96.
Attaix D, Combaret L, Bechet D, Taillandier D. Role of the ubiquitin-proteasome pathway in muscle atrophy in cachexia. Curr Opin Support Palliat Care. 2008;2(4):262–6.
Murphy KT, Chee A, Trieu J, Naim T, Lynch GS. Inhibition of the renin-angiotensin system improves physiological outcomes in mice with mild or severe cancer cachexia. Int J Cancer. 2013;133(5):1234–46.
Adigun AQ, Ajayi AA. The effects of enalapril-digoxin-diuretic combination therapy on nutritional and anthropometric indices in chronic congestive heart failure: preliminary findings in cardiac cachexia. Eur J Heart Fail. 2001;3(3):359–63.
Schanze N, Springer J. Evidence for an effect of ACE inhibitors on cancer cachexia. J Cachex Sarcopenia Muscle. 2012;3(2):139.
Mantovani G, Madeddu C, Maccio A, Gramignano G, Lusso MR, Massa E, et al. Cancer-related anorexia/cachexia syndrome and oxidative stress: an innovative approach beyond current treatment. Cancer Epidemiol Biomark Prev. 2004;13(10):1651–9.
Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185(6):1083–95.
Attaix D, Combaret L, Tilignac T, Taillandier D. Adaptation of the ubiquitin-proteasome proteolytic pathway in cancer cachexia. Mol Biol Rep. 1999;26(1–2):77–82.
Lazarus DD, Destree AT, Mazzola LM, McCormack TA, Dick LR, Xu B, et al. A new model of cancer cachexia: contribution of the ubiquitin-proteasome pathway. Am J Physiol. 1999;277(2 Pt 1):E332–41.
Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer. 2005;93(7):774–80.
Combaret L, Ralliere C, Taillandier D, Tanaka K, Attaix D. Manipulation of the ubiquitin-proteasome pathway in cachexia: pentoxifylline suppresses the activation of 20S and 26S proteasomes in muscles from tumor-bearing rats. Mol Biol Rep. 1999;26(1–2):95–101.
Russell ST, Siren PM, Siren MJ, Tisdale MJ. Attenuation of skeletal muscle atrophy in cancer cachexia by D-myo-inositol 1,2,6-triphosphate. Cancer Chemother Pharmacol. 2009;64(3):517–27.
Busquets S, Garcia-Martinez C, Alvarez B, Carbo N, Lopez-Soriano FJ, Argiles JM. Calpain-3 gene expression is decreased during experimental cancer cachexia. Biochim Biophys Acta. 2000;1475(1):5–9.
Zhang L, Tang H, Kou Y, Li R, Zheng Y, Wang Q, et al. MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J Cancer Res Clin Oncol. 2013;139(7):1105–15.
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18(1):39–51.
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell. 2004;117(3):399–412.
Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol. 2006;291(3):R674–83.
Xu H, Crawford D, Hutchinson KR, Youtz DJ, Lucchesi PA, Velten M, et al. Myocardial dysfunction in an animal model of cancer cachexia. Life Sci. 2011;88(9–10):406–10.
Mastrocola R, Reffo P, Penna F, Tomasinelli CE, Boccuzzi G, Baccino FM, et al. Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radic Biol Med. 2008;44(4):584–93.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–8.
Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. 2007;117(12):3940–51.
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF-1/PI3K/Akt Pathway Prevents Expression of Muscle Atrophy-Induced Ubiquitin Ligases by Inhibiting FOXO Transcription Factors. Mol Cell. 2004;14(3):395–403.
Fontes-Oliveira CC, Busquets S, Toledo M, Penna F, Aylwin MP, Sirisi S, Silva AP, Orpi M, Garcia A, Sette A et al.: Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: Altered energetic efficiency? Biochim Biophys Acta. 2013;1830(3):2770-8.
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metab. 2007;6(6):472–83.
Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 2012;26(3):987–1000.
Liu CM, Yang Z, Liu CW, Wang R, Tien P, Dale R, et al. Effect of RNA oligonucleotide targeting Foxo-1 on muscle growth in normal and cancer cachexia mice. Cancer Gene Ther. 2007;14(12):945–52.
Asp ML, Tian M, Wendel AA, Belury MA. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int J Cancer. 2010;126(3):756–63.
Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med. 2008;86(10):1113–26.
Cong H, Sun L, Liu C, Tien P. Inhibition of atrogin-1/MAFbx expression by adenovirus-delivered small hairpin RNAs attenuates muscle atrophy in fasting mice. Hum Gene Ther. 2011;22(3):313–24.
Williams A, Sun X, Fischer JE, Hasselgren PO. The expression of genes in the ubiquitin-proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery. 1999;126(4):744–9. discussion 749–750.
Bossola M, Muscaritoli M, Costelli P, Bellantone R, Pacelli F, Busquets S, et al. Increased muscle ubiquitin mRNA levels in gastric cancer patients. Am J Physiol Regul Integr Comp Physiol. 2001;280(5):R1518–23.
Bossola M, Muscaritoli M, Costelli P, Grieco G, Bonelli G, Pacelli F, et al. Increased muscle proteasome activity correlates with disease severity in gastric cancer patients. Ann Surg. 2003;237(3):384–9.
DeJong CH, Busquets S, Moses AG, Schrauwen P, Ross JA, Argiles JM, et al. Systemic inflammation correlates with increased expression of skeletal muscle ubiquitin but not uncoupling proteins in cancer cachexia. Oncol Rep. 2005;14(1):257–63.
Williams JP, Phillips BE, Smith K, Atherton PJ, Rankin D, Selby AL, et al. Effect of tumor burden and subsequent surgical resection on skeletal muscle mass and protein turnover in colorectal cancer patients. Am J Clin Nutr. 2012;96(5):1064–70.
Khal J, Hine AV, Fearon KC, Dejong CH, Tisdale MJ. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int J Biochem Cell Biol. 2005;37(10):2196–206.
Jagoe RT, Redfern CP, Roberts RG, Gibson GJ, Goodship TH. Skeletal muscle mRNA levels for cathepsin B, but not components of the ubiquitin-proteasome pathway, are increased in patients with lung cancer referred for thoracotomy. Clin Sci. 2002;102(3):353–61.
Op den Kamp CM, Langen RC, Minnaard R, Kelders MC, Snepvangers FJ, Hesselink MK, et al. Pre-cachexia in patients with stages I-III non-small cell lung cancer: systemic inflammation and functional impairment without activation of skeletal muscle ubiquitin proteasome system. Lung Cancer. 2012;76(1):112–7.
Smith IJ, Aversa Z, Hasselgren PO, Pacelli F, Rosa F, Doglietto GB, et al. Calpain activity is increased in skeletal muscle from gastric cancer patients with no or minimal weight loss. Muscle Nerve. 2011;43(3):410–4.
Schmitt TL, Martignoni ME, Bachmann J, Fechtner K, Friess H, Kinscherf R, et al. Activity of the Akt-dependent anabolic and catabolic pathways in muscle and liver samples in cancer-related cachexia. J Mol Med. 2007;85(6):647–54.
Rhoads MG, Kandarian SC, Pacelli F, Doglietto GB, Bossola M. Expression of NF-kappaB and IkappaB proteins in skeletal muscle of gastric cancer patients. Eur J Cancer. 2010;46(1):191–7.
Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010;2(1):1.
Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA, et al. Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcriptome in sequential human muscle biopsies. Clin Cancer Res. 2012;18(10):2817–27.
Bechet D, Tassa A, Taillandier D, Combaret L, Attaix D. Lysosomal proteolysis in skeletal muscle. Int J Biochem Cell Biol. 2005;37(10):2098–114.
Sakuma K, Yamaguchi A. Sarcopenia and cachexia: the adaptations of negative regulators of skeletal muscle mass. J Cachex Sarcopenia Muscle. 2012;3(2):77–94.
Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4(4):524–6.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007;6(6):458–71.
Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280(21):20814–23.
Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011;25(1):99–110.
McClung JM, Judge AR, Powers SK, Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol. 2010;298(3):C542–9.
Tessitore L, Costelli P, Baccino FM. Pharmacological interference with tissue hypercatabolism in tumour-bearing rats. Biochem J. 1994;299(Pt 1):71–8.
Tessitore L, Costelli P, Bonetti G, Baccino FM. Cancer cachexia, malnutrition, and tissue protein turnover in experimental animals. Arch Biochem Biophys. 1993;306(1):52–8.
Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, et al. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol. 2013;182(4):1367–78.
Schersten T, Lundholm K. Lysosomal enzyme activity in muscle tissue from patients with malignant tumor. Cancer. 1972;30(5):1246–51.
Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24(50):7455–64.
Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S, et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci. 2011;342(2):143–7.
Eley HL, Russell ST, Tisdale MJ. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem J. 2007;407(1):113–20.
Robert F, Mills JR, Agenor A, Wang D, DiMarco S, Cencic R, et al. Targeting protein synthesis in a Myc/mTOR-driven model of anorexia-cachexia syndrome delays its onset and prolongs survival. Cancer Res. 2012;72(3):747–56.
Eley HL, Skipworth RJ, Deans DA, Fearon KC, Tisdale MJ. Increased expression of phosphorylated forms of RNA-dependent protein kinase and eukaryotic initiation factor 2alpha may signal skeletal muscle atrophy in weight-losing cancer patients. Br J Cancer. 2008;98(2):443–9.
Lagirand-Cantaloube J, Cornille K, Csibi A, Batonnet-Pichon S, Leibovitch MP, Leibovitch SA. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS One. 2009;4(3):e4973.
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin Jr AS. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289(5488):2363–6.
He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest. 2013;123(11):4821–35.
Emery PW. Cachexia in experimental models. Nutrition. 1999;15(7–8):600–3.
Fearon KCH. The 2011 ESPEN Arvid Wretlind lecture: Cancer cachexia: The potential impact of translational research on patient-focused outcomes. Clin Nutr. 2012;31(5):577–82.
Fearon K, Arends J, Baracos V: Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol. 2013;10(2):90-9. doi: 10.1038/nrclinonc.2012.209. Epub 4 Dec 2012.
Baracos VE, Reiman T, Mourtzakis M, Gioulbasanis I, Antoun S. Body composition in patients with non-small cell lung cancer: a contemporary view of cancer cachexia with the use of computed tomography image analysis. Am J Clin Nutr. 2010;91(4):1133S–7S.
Knox LS, Crosby LO, Feurer ID, Buzby GP, Miller CL, Mullen JL. Energy expenditure in malnourished cancer patients. Ann Surg. 1983;197(2):152–62.
Baracos VE. Clinical Trials of Cancer Cachexia Therapy, Now and Hereafter. J Clin Oncol. 2013;31:1257–8.
Punzi T, Fabris A, Morucci G, Biagioni P, Gulisano M, Ruggiero M, et al. C-reactive protein levels and vitamin d receptor polymorphisms as markers in predicting cachectic syndrome in cancer patients. Mol Diagn Ther. 2012;16(2):115–24.
Deans DA, Tan BH, Ross JA, Rose-Zerilli M, Wigmore SJ, Howell WM, et al. Cancer cachexia is associated with the IL10–1082 gene promoter polymorphism in patients with gastroesophageal malignancy. Am J Clin Nutr. 2009;89(4):1164–72.
Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, Deans DA, et al. P-selectin genotype is associated with the development of cancer cachexia. EMBO Mol Med. 2012;4(6):462–71.
Gioulbasanis I, Vlachostergios PJ, Papandreou CN. Selecting for predisposition to cancer cachexia. EMBO Mol Med. 2012;4(6):451–2.
Johns N, Stephens NA, Preston T. Muscle protein kinetics in cancer cachexia. Curr Opin Support Palliat Care. 2012;6(4):417–23.
Thoresen L, Frykholm G, Lydersen S, Ulveland H, Baracos V, Prado CM, et al. Nutritional status, cachexia and survival in patients with advanced colorectal carcinoma. Different assessment criteria for nutritional status provide unequal results. Clin Nutr. 2013;32(1):65–72.
Tan BH, Birdsell LA, Martin L, Baracos VE, Fearon KC. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin Cancer Res. 2009;15(22):6973–9.
Gordon JN, Trebble TM, Ellis RD, Duncan HD, Johns T, Goggin PM. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut. 2005;54(4):540–5.
Maccio A, Madeddu C, Mantovani G. Current pharmacotherapy options for cancer anorexia and cachexia. Expert Opin Pharmacother. 2012;13(17):2453–72.
Reid J, Mills M, Cantwell M, Cardwell CR, Murray LJ, Donnelly M. Thalidomide for managing cancer cachexia. Cochrane Database Syst Rev. 2012;4:CD008664.
Jatoi A, Dakhil SR, Nguyen PL, Sloan JA, Kugler JW, Rowland Jr KM, et al. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: results from N00C1 from the North Central Cancer Treatment Group. Cancer. 2007;110(6):1396–403.
Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G, et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol. 2008;6(1):18–25.
Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther. 2011;11(12):1663–8.
Prado CM, Bekaii-Saab T, Doyle LA, Shrestha S, Ghosh S, Baracos VE, et al. Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: selumetinib in patients with cholangiocarcinoma. Br J Cancer. 2012;106(10):1583–6.
Norden DM, Devine R, McCarthy DO, Wold LE. Storage conditions and passages alter IL-6 secretion in C26 adenocarcinoma cell lines. MethodsX. 2015;2:53–8.
Vanchieri C. Cachexia in cancer: is it treatable at the molecular level? J Natl Cancer Inst. 2010;102(22):1694–7.
Shyu KG, Lu MJ, Wang BW, Sun HY, Chang H. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur J Clin Investig. 2006;36(10):713–9.
George I, Bish LT, Kamalakkannan G, Petrilli CM, Oz MC, Naka Y, et al. Myostatin activation in patients with advanced heart failure and after mechanical unloading. Eur J Heart Fail. 2010;12(5):444–53.
Heineke J, Auger-Messier M, Xu J, Sargent M, York A, Welle S, et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121(3):419–25.
Lee SJ, Glass DJ. Treating cancer cachexia to treat cancer. Skelet Muscle. 2011;1(1):2.
Tisdale MJ. Re: Wieland BM, et al. Is there a human homologue to the murine proteolysis-inducing factor? Clin Cancer Res. 2008;14(7):2245–6.
Tisdale MJ. The ubiquitin-proteasome pathway as a therapeutic target for muscle wasting. J Support Oncol. 2005;3(3):209–17.
Jatoi A, Alberts SR, Foster N, Morton R, Burch P, Block M, et al. Is bortezomib, a proteasome inhibitor, effective in treating cancer-associated weight loss? Preliminary results from the North Central Cancer Treatment Group. Support Care Cancer. 2005;13(6):381–6.
Baldwin C, Spiro A, Ahern R, Emery PW. Oral nutritional interventions in malnourished patients with cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2012;104(5):371–85.
Argiles JM, Busquets S, Lopez-Soriano FJ, Costelli P, Penna F. Are there any benefits of exercise training in cancer cachexia? J Cachex Sarcopenia Muscle. 2012;3(2):73–6.
Lundholm K, Korner U, Gunnebo L, Sixt-Ammilon P, Fouladiun M, Daneryd P, et al. Insulin treatment in cancer cachexia: effects on survival, metabolism, and physical functioning. Clin Cancer Res. 2007;13(9):2699–706.
Lundholm K, Daneryd P, Korner U, Hyltander A, Bosaeus I. Evidence that long-term COX-treatment improves energy homeostasis and body composition in cancer patients with progressive cachexia. Int J Oncol. 2004;24(3):505–12.
Acknowledgments
We thank Prof. J. Kleeff and Prof. K.P. Jannsen (Department of Surgery, Klinikum Rechts der Isar, Technische Universität München, Munich Germany) for their advice regarding this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TCM, MM, HF, JB, OP conception and design. TCM, JB literature search and review. TCM, MM, HF, JB, OP interpretation of data. TCM manuscript draft and writing of manuscript. TCM, MM, HF, JB, OP revision of manuscript and approval of final version.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mueller, T.C., Bachmann, J., Prokopchuk, O. et al. Molecular pathways leading to loss of skeletal muscle mass in cancer cachexia – can findings from animal models be translated to humans?. BMC Cancer 16, 75 (2016). https://doi.org/10.1186/s12885-016-2121-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-016-2121-8