
Abstract
Background
The development of androgen resistance is a major limitation to androgen deprivation treatment in prostate cancer. We have developed an in vitro model of androgen-resistance to characterise molecular changes occurring as androgen resistance evolves over time. Our aim is to understand biological network profiles of transcriptomic changes occurring during the transition to androgen-resistance and to validate these changes between our in vitro model and clinical datasets (paired samples before and after androgen-deprivation therapy of patients with advanced prostate cancer).
Methods
We established an androgen-independent subline from LNCaP cells by prolonged exposure to androgen-deprivation. We examined phenotypic profiles and performed RNA-sequencing. The reads generated were compared to human clinical samples and were analysed using differential expression, pathway analysis and protein-protein interaction networks.
Results
After 24 weeks of androgen-deprivation, LNCaP cells had increased proliferative and invasive behaviour compared to parental LNCaP, and its growth was no longer responsive to androgen. We identified key genes and pathways that overlap between our cell line and clinical RNA sequencing datasets and analysed the overlapping protein-protein interaction network that shared the same pattern of behaviour in both datasets. Mechanisms bypassing androgen receptor signalling pathways are significantly enriched. Several steroid hormone receptors are differentially expressed in both datasets. In particular, the progesterone receptor is significantly differentially expressed and is part of the interaction network disrupted in both datasets. Other signalling pathways commonly altered in prostate cancer, MAPK and PI3K-Akt pathways, are significantly enriched in both datasets.
Conclusions
The overlap between the human and cell-line differential expression profiles and protein networks was statistically significant showing that the cell-line model reproduces molecular patterns observed in clinical castrate resistant prostate cancer samples, making this cell line a useful tool in understanding castrate resistant prostate cancer. Pathway analysis revealed similar patterns of enriched pathways from differentially expressed genes of both human clinical and cell line datasets. Our analysis revealed several potential mechanisms and network interactions, including cooperative behaviours of other nuclear receptors, in particular the subfamily of steroid hormone receptors such as PGR and alteration to gene expression in both the MAPK and PI3K-Akt signalling pathways.
Similar content being viewed by others

Background
Prostate cancer (PCa) is among the most commonly diagnosed diseases in males, and remains a leading cause of death in developed countries [1]. In Australia, PCa is the most commonly diagnosed cancer and accounted for approximately 13 % of all cancer-related deaths in males in 2010 [2, 3]. PCa tumour growth is initially dependent on androgens as documented by Huggins as early as 1941 [4], making androgen deprivation therapy (ADT) the first line treatment. However patients often ultimately develop an androgen independent state of PCa, often referred to as Castrate Resistant Prostate Cancer (CRPC) and there are no effective treatments for this state of PCa.
Androgens act through the androgen receptor (AR) signalling pathway. A review by Feldman [5] details five broad mechanisms through which PCa cells can survive despite low levels of serum testosterone in CRPC. Three out of the five mechanisms involve AR signalling, where in the absence of serum testosterone, AR continues to play an active role in CRPC through adrenal testosterone, increased AR expression level (AR amplification), AR mutation where other steroid hormones (such as progesterone or oestrogen) or mutated co-regulators activate AR and “outlaw” AR where AR becomes ligand independent, for example through alternative splicing. The other two mechanisms bypass AR altogether, and CRPC cells survive through alternative pathways, such as through up-regulation of oncogenes that block signals for cell apoptosis and cause cell proliferation.
The establishment of an in vitro model of CRPC is crucial for the study of the progression into advanced stage PCa. Previous studies have used the androgen-sensitive cell line LNCaP in long-term culture in androgen deprived conditions. These long-term cultures were carried out ranging from 2 months up to 24 months during which time androgen resistance develops in these cells [6–9].
The question remains whether these in vitro studies reflect biological features in human tumours, a question addressed in this current work. In our experiments, we study the various CRPC mechanisms using existing human tumour datasets [10] and in vitro LNCaP cell line model through computational methods of RNA sequencing expression, differential expression and network analysis. Data from transcriptomic profiling of patients [10] receiving ADT (LHRH agonists with anti-androgen flare protection [11]) for approximately 22 weeks were compared with our cell line model to determine what molecular changes were common to the two datasets, and to establish the suitability of our model system for studying drivers of developing androgen insensitivity in vivo.
Methods
LNCaP cell line and reagents
The human PCa cell line LNCaP was obtained from American Type Culture Collection (ATCC) (Manassas, Virginia, USA). Cells were maintained and propagated as monolayer cultures in RPMI 1640 medium (Life Technologies Corporation) with 10 % foetal bovine serum (FBS) (Thermo Scientific), and 100 units/mL penicillin and 100 μg/mL streptomycin (Life Technologies Corporation).
In vitro androgen independent model
In vitro CRPC models were established by prolonged cultures of androgen-sensitive LNCaP cells (parental). We have generated cells grown under (i) a control condition for parental LNCaP, in FBS; (ii) media with charcoal-stripped FBS which removes low molecular weight hormones including steroid, thyroid and peptide hormones (CS-FBS, androgen-deprived) (Fig. 1). Cells that were grown in androgen-deprived condition are referred to as LNCaP androgen independent (LNCaP AI) cells.

In vitro CRPC cells workflow. All conditions are replicated in triplicate so that each condition has three independent biological replicates
Cell viability assay
Trypan blue dye exclusion was performed to examine cell viability of cell lines. Routine cell harvesting was performed and cell suspension was diluted (1:1) with 0.1 % (w/v) trypan blue dye (Sigma Aldrich) in dH2O and transferred (20 μL) to a haemocytometer for counting, using an inverted microscope (Model CK2, Olympus Optical Co. Ltd, Japan). Total of viable and non-viable cell numbers were counted by trypan blue dye exclusion.
Cell proliferation assay
Relative cell numbers were measured by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide or MTT colorimetric assays. Parental cells were grown in phenol-red free CS-FBS media 3 days prior to the start of the experiments. Cells were plated on 96-well plates in phenol-red free media + CS-FBS. After overnight attachment, cells were treated with 1–10 nM DHT or 1–10 μM bicalutamide for 6 days. Cell proliferation was examined by addition of MTT to the assay plate and the absorbance read at 590 nm, reference filter 620 nm.
RNA isolation
RNA was isolated from cells in triplicate using the Rneasy Mini Kit (Qiagen Pty Ltd.) according to manufacturer’s instructions. RNA quantity was assessed using a NanoDrop 2000 UV–vis Spectrophotometer (Thermo Scientific) at A260nm and RNA integrity was determined using the A260nm/A280nm ratio. For RNA sequencing, RNA was checked for yield and quality using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.).
Reverse-transcript PCR and quantitative PCR (qPCR)
RNA was extracted from parental LNCaP and subline cells as described above. cDNA synthesis were performed using M-MuL-V kit (Life Technologies Corporation). cDNA samples were then analysed using AR (assay ID Hs00171172_m1) and 18S (assay ID Hs99999901_s1) (as control) Taqman gene expression assays (Applied Biosystems, Life Technologies Corporation). PCR amplification was performed in a 25 μL final volume (total 54 ng cDNA per reaction) using 7500 Real-time PCR System (Applied Biosystems, Life Technologies Corporation). mRNA expression of AR was normalized in relation to the control 18S expression. Data are expressed as fold difference to parental LNCaP cell line.
Lysate extraction and western blotting
Modified radioimmunoprecipitation (RIPA) buffer was used to extract proteins from the cell. Medium was removed from cells and cells were washed twice with ice-cold PBS before addition of ice-cold RIPA buffer containing 1× Complete Mini EDTA-free protease inhibitor tablet (Roche Diagnostics). Protein concentration of the whole cell lysates was determined using the Bradford assay [12]. Proteins were separated by SDS-PAGE. Protein bands were then transferred to nitrocellulose paper and incubated with 1/200 diluted AR antibody (N-20) [Santa Cruz Biotechnology] and peroxidase conjugated antibody respectively. Peroxidase linked antibody was purchased from Amersham™ (GE Healthcare Biosciences). β-actin levels were used as a loading control. Protein bands were visualized after chemiluminescent reaction.
Immunocytochemistry
Preparation of cover slips
Cover slips were positioned in a sterile beaker and were immersed in ice-cold 100 % (v/v) methanol under aseptic conditions. The beaker was placed in a container filled with ice and left in the fume hood under UV light for 2 h to sterilise. The cover slips were allowed to dry and were then placed into each well of the 6-well plate.
Cells were passaged by trypsinisation. Cell suspensions were added to prepared 6-well plate (with cover slip in each well) at a concentration of 1 × 105 cells/well. Cells were allowed to grow at 37 °C/5 % CO2 in a humidified incubator to a confluence of 50–70 %, with addition of fresh media if needed.
Immunostaining
When cells had reached confluence, the old media were aspirated from each well. Coverslips were washed with PBS buffer for 5 min and then were fixed in ice-cold 100 % (v/v) methanol for 10 min at room temperature. Cells were permeabilised in PBS containing 0.1 % (v/v) Triton-X 100 for 5 min. The cover slips were washed twice with PBS for 5 min. Immunohistochemical analysis was performed using the labelled streptavidin/biotin-based LSAB + ™ (Labeled Streptavidin Biotin) system/HRP kit (DAKO Australia) according to the manufacturer’s protocol, with minor alterations. All incubations were carried out at room temperature. Primary antibody monoclonal mouse anti-human PGR antibody (clone A9621A, R&D systems) at working concentration of 1 ug/mL in an antibody diluent solution (DAKO Australia) was applied to each slide for 1 hour. This was followed by sequential incubation of fixed cells with anti-goat/rabbit/mouse biotinylated-link antibody and peroxidase-labelled streptavidin. PGR protein expression was visualised by incubation with 3,3’-diaminobenzidine chromogen solution (DAB+ substrate buffer), yielding a brown end-product. Fixed cells were counterstained with haematoxylin. As a negative control, the fixed cells were incubated with isotype antibody IgG2a.
Cell migration and invasion assays
Migration assays were performed in BIOCOAT™ control microporous membrane filter inserts while invasion assays were performed on Matrigel matrix-treated polyethylene terephthalate (PET) membrane filter inserts in 24-well tissue culture plates (BD Biosciences, Australia), as described previously [13]. Briefly, the BIOCOAT™ inserts are 6.4 mm in diameter, and the pore size on the membrane is 8 μm. Cells were washed once with versene, detached at 80–90 % confluence with 0.05 % trypsin/EDTA, and resuspended in serum-free media. The inserts were incubated with serum-free media at 37 °C for 2 h to rehydrate. Media containing 10 % FCS (as a chemoattractant) was added to the lower wells and a 500 μL cell suspension was added to the inserts at a density of 5 × 104 cells/insert. Migration across the membrane was allowed to proceed at 37 °C 5 % CO2 for 48 hours. Cells that did not migrate through the membrane were removed using cotton swabs, and cells that migrated through the membrane filters were fixed with 100 % v/v methanol and stained with 0.05 % v/v crystal violet dye (Sigma-Aldrich, Australia). The membranes were carefully removed from the insert using a scalpel blade and mounted onto glass slides. The number of migrated cells per insert (10 fields were chosen from each insert) was counted using the M2 program of the Micro-Computer Imaging Device-assisted image analysis program (MCID, Imaging Research, Inc., St. Catharine’s; Ontario, Canada) under light microscopy at magnification ×200. All experiments were repeated in triplicate on each of three separate occasions.
LNCaP RNA sequencing
LNCaP and LNCaP-AI RNA samples were sequenced on an Illumina platform (Illumina HiSeq2000) by the Australian Genome Research Facility (AGRF), Melbourne, Australia. CASAVA1.8 pipeline was used to generate the sequence data. The sequence reads were processed through a quality control pipeline (FastQC, SolexaQA) (investigating quality matrices such as presence of ambiguous bases, adaptor contamination, PCR duplicates, GC content and sequence complexity) with required quality score > Q30 for all reads.
Human clinical data
The RNA reads from 7 patients taken before (Hormone naïve) and after (Hormone resistant, defined as two consecutive rise of PSA more than 10 %) androgen-deprivation treatment (ADT) (approximately 22 weeks) were obtained from a study conducted by Rajan et al. [10] on the effects of ADT on advanced PCa. mRNA reads, obtained by next gen sequencing techniques were reanalysed using the protocol described below.
Computational methods
We have used standard methods for RNA sequencing differential expression analysis. Tophat v2.0.9 [14] was used to align the RNA sequencing reads using hg19 as the reference genome and EdgeR v3.4.2 [15] was used for differential expression analysis.
For our protein to protein network analysis, we obtained the network for homo sapiens from PINA2 interaction resource [16]. The gene, UBC, which has >5000 interactions in this dataset was removed from the analysis. We constructed a protein interaction network by taking the set of genes differentially expressed in both datasets, and collecting all protein interactions involving the products of these genes by querying the human interactome with Uniprot Accession numbers obtained from Biomart.org. The resulting network was pruned to remove proteins with a degree of 1, such that a protein not in our original list of commonly differentially expressed genes was only retained in the network if it interacted with at least two proteins encoded by the query set.
We also used machine learning to predict gene regulatory network using GENIE3 [17]. GENIE3 uses random forests for regression trees to compute an importance measure of the relationship between the predictor variables and the output variable. Modelling gene regulatory network inference as a machine learning problem, the expression level of each gene, computed using cuffnorm [14], is the output variable and the expression levels of transcription factors are the predictor variables.
All statistical tests were carried out in R using the hypergeometric distribution test function.
Results
Characterisation of androgen-deprived LNCaP subline cells (LNCaP-AI)
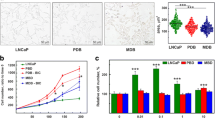
LNCaP cells were grown in culture under androgen-deprived conditions for 24 weeks (LNCaP-AI). The viability of cells in culture was examined regularly and LNCaP-AI cells initially showed poor growth and proliferation after growth in androgen-deprived culture. Over a period of time, however, cells adapted and started to grow vigorously (data not shown). Dihydrotestosterone (DHT) dose–response stimulation of cell proliferation was performed for parental LNCaP and LNCaP-AI after prolonged culture of 24 weeks to determine androgen responsiveness. These assays established that LNCaP-AI cells lost androgen-responsiveness after 24 weeks of culture when compared to parental LNCaP cells that were grown in parallel, DHT (concentration from 0.1 to 10 nM) not being able to stimulate increased proliferation in LNCaP-AI (Fig. 2a).

Androgen responsiveness of LNCaP and LNCaP-AI cells. a Androgen responsiveness of LNCaP-AI cells was lost by 24 weeks of prolonged culture compared to parental LNCaP cells. Data are shown as % cell proliferation of DHT treated vs. no DHT treatment condition. For this proliferation assay, cells were treated with different concentrations of DHT for a period of 6 days. b No difference in cell migration was seen between parental and subline cells. However, LNCaP-AI cells were more invasive (c) compared to parental LNCaP cells. (N = 3, error bars = SEM, * indicates P < 0.05). Picture inset underneath the graphs are representative images of cell migration and invasion (cells are stained purple)
AR expression and activation
We examined other phenotypic characteristics and observed that LNCaP-AI cells also displayed increased cell invasion after 24 weeks of prolonged culture compared to parental control cells, using Boyden chamber cell invasion assays (Fig. 2b and c), consistent with a more aggressive phenotype. AR gene (qPCR) and protein (Western Blot) (Fig. 3a and b, respectively) expression in LNCaP-AI cells was not statistically significantly (P > 0.05) different to parental LNCaP cells. This is consistent with the fact that increased AR expression is not the sole determinant of initiation of PCa or development of hormone refractory PCa [18].

AR expression in LNCaP and LNCaP-AI. a AR gene and b Protein expression (densitometric analysis of AR corrected for β-actin expressed in AU [arbitrary units]) of LNCaP and LNCaP-AI cells at 24 weeks of culture. (N = 3, error bars = SEM)
Given that our cell line model demonstrates a phenotype consistent with androgen insensitivity after 24 weeks of growth in androgen deprived culture conditions, we wished to establish how well our model reproduces the molecular phenotype seen in androgen resistant human tumours. To do this, we compared the results of our RNA sequencing of sensitive and insensitive cells, with a previously published study of gene expression in human tumours before and after treatment with androgen deprivation therapy. To ensure the results were comparable, we re-analysed the human data using the same analysis protocol we used for our own RNA sequencing data (see Methods section).
The AR was not differentially expressed in either the human tumour samples or the cell line model (Fig. 4). However genes, such as KLK3 and TMPRSS2 that are normally uniquely up regulated by AR are down regulated as shown in Fig. 4 suggesting that AR is not actively regulating these targets.

Expression levels of AR regulated target genes. Green nodes indicate under expressed genes, whereas red nodes represent over expressed genes; edges are coloured to indicate the expected direction of regulation (red edges indicate positive regulation, whereas green edges indicate inhibition). Genes shown in grey are not significantly differentially expressed. a AR regulated genes in human tumour samples; b AR regulated genes in LNCaP-AI cells
We also examined expressed AR Isoforms AR-001, AR-002, AR-003, AR-004 (AR-V7) [19], AR-005 and AR-201 to determine if differential expression of any of these isoforms might underpin the androgen insensitive phenotype in either our cell lines or the human tumours (Table 1). We found that none of the isoforms were significantly differentially expressed either after treatment in patient samples, or in our cell line model. Interestingly, several isoforms were detectable in human tumour data both before and after treatment, and in both time points of our experimental system, and transcripts show great variation in composition of functional domains (Table 1).
Common differentially expressed genes are found in human PCa and LNCaP models
We used a published clinical dataset [10] to determine whether our cell line model of androgen insensitivity displays molecular features in common with human disease. We found 213 genes were differentially expressed in both experiments. This highlights that while there are definite differences in the molecular phenotypes of our model and the human tumour data, our model none-the-less recapitulates molecular features found in advanced human disease. We also examined the pathways that are enriched for differentially expressed genes in each experiment, and found that differential expression in both datasets converged at the pathway level (full results Additional file 1: Table S1 and S2); in particular, two related pathways, MAPK and PI3K signalling are both strongly implicated by the differentially expressed genes of both datasets. Previous reports adopting disease-associated gene network and pathway analyses in PCa have revealed novel regulatory mechanisms and were more powerful than the analysis of gene expression level alone [20–24].
Analysis of protein network overlap between in the human PCa and LNCaP models
In order to explore the mechanisms captured in our cell line model and in common with those in the human tumour data, we focused on the set of 213 genes that were differentially expressed in both datasets and first examined protein interactions among the proteins encoded by these genes (Fig. 5). In particular, the presence of two up-regulated nuclear receptor transcription factors in this network (PGR and NR2F1) suggests that these transcription factors may play a role in the androgen insensitive state. As the functional effects of differential expression propagate through the interaction partners of proteins, we expanded our protein interaction network to include interaction partners of the 213 commonly differentially expressed genes in order to capture a broader set of proteins whose functions are likely to be affected by these changes. The initial network expansion added 1700 new interacting proteins, 80 % of which interacted with only one protein from our query set. We subsequently pruned this network in order to increase the likelihood of identifying proteins through which the functional effects of altered gene expression are likely to propagate: we removed proteins with a degree of 1 in this network, and insisted that all proteins interacted with at least two from our query set of 213. This resulted in a network of 492 proteins with 1062 interactions, which we refer to as our disrupted network. While genes differentially expressed in both experiments encode only 170 proteins in this network (as some of the 213 lack known interactions), a further 85 correspond to genes disrupted in either one or the other experiment, such that 45 % of all proteins captured in this network have evidence for significant differential expression in PCa either in human tumours or in our cell line model.

Protein interactions within the set of 213 genes differentially expressed in both experimental datasets. AR has been included in this network for reference. Red nodes represent genes with increased expression in the resistant state, whereas green genes have lower expression. AR itself is not differentially expressed
In order to determine the likely effects of this disrupted network on the development of androgen resistance in our model, we performed a functional analysis looking at enriched Gene Ontology terms and signalling pathways. Analysis of the molecular functions enriched in this network revealed two very strong functional signatures, related to transcription factor binding (58 genes, corrected P value 1.9 × 10−19, including steroid hormone receptor binding with a corrected p value of 4.2 × 10−8), and protein kinase activity (55 genes, corrected p value 2.7 × 10−14). This suggested that our disrupted network represents two broad adaptive mechanisms that may be at play in both our model and the human tumour data: alteration of transcriptional regulation in response to loss of AR regulation (seen in Fig. 4); and altered signalling driving proliferation and inhibiting cell death. To explore both these mechanisms, we (i) performed analysis of the regulatory impact of transcription factors, and (ii) performed pathway analysis of the disrupted network .
-
(i)
Regulatory impact of nuclear receptors in androgen insensitive tumours and cells
In the human data, 15 of 48 known nuclear receptors are differentially expressed and of the 8 steroid hormone receptors, four are differentially expressed (Table 2). In our cell line data set, 11 nuclear receptors are differentially expressed (Table 2). It has previously been reported that nuclear receptors (NRs), particularly the sub family of steroid hormone receptors, with a similar structure and binding motif, may provide some functional redundancy. Thus, we hypothesize that other nuclear receptors may be up regulated in response to loss of AR signalling, and compensate for the loss of gene regulation by AR in androgen insensitive PCa.
We performed a computational analysis to predict regulatory relationships between NRs and potential target genes. To do this, we use the method Genie3 [17], which uses random forests for regression trees to compute an importance measure for the relationship between the predictor variables (here, the expression level of NR transcription factors) and the output variables (expression levels of all other genes). Because individual predictions of association between transcription factors and targets based on expression data are likely to be noisy and error prone [25], we do not attempt to use these networks to infer mechanism; rather we look for strong patterns of shifting influence as captured by the broad-scale loss or gain of targets with high importance measures (the top ten transcription factors ranked by increasing size of inferred regulatory networks are shown for human tumour data in Table 3, and cell line data in Table 4).
Most notably, the progesterone receptor (PGR), showed the largest increase in inferred network size of all nuclear receptors in both the human and cell line data (we confirmed PGR protein expression via immunocytochemistry (Fig. 6)), suggesting that this transcription factor may be assuming a regulatory role that in part compensates for the loss of AR regulation. It should be noted that these networks are derived from relationships in the expression data, and are not influenced by the size of the interaction networks available for these transcription factors.

PGR protein expression in prostate cancer cell line, LNCaP (Panel a) and LNCaP-AI (Panel b). Cells were grown, fixed and then stained for PGR (A1, B1) and control antibody (A2, B2). The localisation of PGR was more predominant in LNCaP-AI than LNCaP in the cytoplasm. PGR expression in the Nucleus is also more prominent in LNCaP-AI cells. Original magnification ×400
-
(ii)
Pathway analysis of the disrupted network
We observed a strong enrichment for protein kinase function in our disrupted network, hinting at adaptive mechanisms operating through signalling pathways to promote proliferation in our androgen insensitive cells. We mapped differentially expressed genes from our disrupted network to KEGG signalling pathways in order to identify mechanisms through which these gene expression changes may affect cellular phenotype. Notably, MAPK and PI3K-Akt signalling both showed a large number of differentially expressed genes (MAPK - cell line 42, human 58; and PI3K-Akt - cell line 65, human 92). Within these pathways, we see some striking patterns in shared disruption. In PI3K-Akt signalling, for example, reactions leading to cell survival, growth and proliferation outcomes are up-regulated in both human and cell line data (see Additional file 1: Figure S2 and S4 where these data are mapped to KEGG pathways). Similarly in MAPK signalling, there is a suggestion of enhanced signalling through to NF-ƙB and c-JUN, which could have an impact on proliferation and anti-apoptotic regulation [26] (see Additional file 1: Figure S1 and S3). Interestingly, both human and cell data indicate that signalling through the MEK-ERK module itself may actually be reduced through a combination of down regulation of MEK and up-regulation of inhibitors of ERK. While this may seem counter intuitive, a recent paper characterising the phosphoproteomic changes in PCa found that signalling through ERK was in fact reduced in androgen-independent PCa [27], consistent with our findings here.
Discussion
Here, we have described an in vitro model of the development of androgen insensitivity in PCa, and compared the characteristics of our cell line model to that of human disease. This cell line model is described here.
Role of AR signalling pathway
In our experiments, we found that AR is not differentially expressed and genes (such as KLK3 and TMPRSS2) that are uniquely up regulated by AR are all down regulated in both the human PCa tumour cells and the LNCaP model as shown Fig. 4. Genes such as CLU, PEG3 etc. that are suppressed by AR are up regulated (except for NDRG1) in human PCa cells, and in the LNCaP model. The difference in AR gene and protein (Fig. 3) expression in LNCaP-AI cells was also not in itself statistically significant, and these results are consistent with loss of AR signalling in both systems.
Clinically, rising levels of prostate specific antigen (PSA) show dependence on AR in CRPC and treatments include directly targeting AR, using AR antagonists [28, 29]. However not all patients respond to AR antagonists, and those who do tend to relapse [28]. This shows that tumour cells in CRPC can continue to survive, bypassing the AR signalling pathway. In our experiment, AR is not active in the LNCaP model and the transcription abundance changes in both tumour and cell line data (Fig. 4) suggest that in at least the human data we compare with here, AR is not actively regulating its normal targets. Alternative pathways bypassing AR signalling are therefore likely to be responsible for the development of androgen insensitivity in our cell line model. This emphasises the clinical relevance of the model we have developed for understanding CRPC that is driven by these kinds of resistance mechanisms. Other genes that have been shown to negatively regulate AR and AR signalling are overexpressed in our experimental model, including Id1 [30] and IFI6 [31]. While these are not likely to be the only mechanisms driving clinical CRPC, a model such as ours that allows for the longitudinal study of these processes will provide valuable insight into CRPC driven by such bypass mechanisms [32].
Role of nuclear receptor super family and the steroid hormone receptor (SHR) subfamily
AR belongs to the steroid receptor family within the nuclear receptor superfamily. Of the 48 nuclear receptors [33] known in the human genome, the sub family of steroid hormone receptors contains ERs (Nr3A1/ESR1, NR3A2/ESR2), ERRs (ESRRA/NR3b1, NR3b2), GR (Nr2c1), MR (Nr3c2), PRs (NR3C3), and AR (NR3C4) [34]. DNA binding domain sequence for AR is very similar to PGR and glucocorticoid receptor (GR) [35], so these genes, that are structurally similar to AR, may be replacing AR regulation of some genes. Given almost all classes of SHR are differentially expressed in our samples (Table 2), a possible explanation is that in the absence of AR activation the other SHRs compensate and become active and are involved in CRPC cell multiplication. GR has already been implicated in CRPC bypassing AR blockade [36] and GR is up regulated in the human data set (Table 2).
In our experiments PGR is differentially expressed and up-regulated (see Table 2) and is also part of the top-scoring protein network (see Fig. 5). Recently, the analysis of samples from over 500 PCa patients who have never been treated with hormone therapy revealed that PGR expression in both areas of prostate tumour epithelium and tumour stroma is an independent prognostic factor of clinical failure [37]. Previously, it has been found that PGR has a negative correlation with AR in hormone refractory PCa [38]. PGR was also found to be increased in CRPC but decreased in localised PCa, although the findings did not reach statistical significance [38]. Our finding reflects this clinical finding that PGR is upregulated in androgen-deprived cells and was expressed negatively to AR. As an oestrogen-regulated gene [39], PGR expression could also be interrelated to the importance of oestrogen receptors (ER) in PCa. The two major subtypes of ER, ERα and ERβ have been previously investigated in PCa [40–42]. The interchanging role between ERβ down-regulation in hormone naïve PCa and ERβ up-regulation in CRPC in matched samples has been documented [43]. Other researchers also speculated that ERβ expression might be mediated by phosphorylated AR at serine 210 (pAR(210)) [41, 43]. Although the role of circulating oestrogen in PCa carcinogenesis is still controversial [44], it may be worth investigating the role of ER and PGR in CRPC. Research into the role of PGR and PCa is still in its early stage and also, the lack of available antibody for all isoforms of PGR hinders advancement in the field. While ER expression is not enriched in our network analysis, their associated relationship with PGR compels the need for further investigation of both nuclear receptors in future study.
NR2F1 and NR2F2, also known as COUP-TFs, are orphan nuclear receptors and they occupy consensus DNA binding sites shared by other nuclear receptors including thyroid hormone receptor, retinoic acid receptor, oestrogen receptor and AR [45–47]. This allows COUP-TFs to modulate transcriptional activities of these nuclear receptors. As well, COUP-TFs are able to exert regulatory control via direct competitive binding to coactivators and corepressors of these nuclear receptors [48]. Although more is known about the role of NR2F2 in PCa [45, 49], both COUP-TF genes, NR2F1 and NR2F2 display nearly 100 % homology in their DNA-binding domain (DBD) and ligand- binding domain (LBD), though no natural ligand has been identified for these COUP-TFs [50]. NR2F2 was found to promote prostate tumourigenesis via inactivation of TGF-β signalling in PTEN-null mice [49]. In contrast, another study found NR2F2 to inhibit PCa cell growth via direct binding to AR preventing androgen-dependent signalling [45]. A previous study has shown that NR2F1 expression was negatively regulated by AR but in the presence of AR antagonist, this gene was positively regulated [51]. It can be speculated that in our model long term growth in androgen-deprived conditions decreases androgen-dependent AR-signalling, and could cause up-regulation of the NR2F1 gene. This may suggest that functions of these COUP-TFs depend on the tumour microenvironment and expression profiles of other nuclear receptors and transcription factors [52]. A balance between the two COUP-TFs may also be important in the control of CRPC progression and further longitudinal studies will be important for determining the shift between these two receptors in CRPC.
AR signalling bypass through MAPK and PI3K-AKT signalling pathways
An effective AR signalling bypass would be through genes that support proliferation and inhibit apoptosis, even in the absence of androgens and AR [5]. In our experiment, there are a number of genes that are differentially expressed in cancer pathways and show complex and varied mechanisms involved in CRPC, making drug targeting challenging.
In the human PCa tumour dataset, BCL-2 is up-regulated and is known to block chemotherapy induced apoptosis [53]. TGF-β signalling may also be responsible for CRPC in the human tumour samples. TGF-β signalling is a pathway that has dual roles, tumour-suppressor and tumour-promoter in cancer depending on the cellular context and clinical stage of the disease [54]. In advanced PCa, TGF-β signalling promotes tumour progression, angiogenesis, invasiveness and epithelial-mesenchyme transition (EMT) [55]. AR and TGF-β signalling mediate EMT and the crosstalk between these two signalling pathways determines apoptotic effects in PCa [56–58].
The MAPK and PI3K-Akt pathways are highly enriched with differentially expressed genes in the cell line and the human tumour datasets. The MAPK and PI3K-Akt pathways are one of the most deregulated growth factor receptor signalling pathways in cancers [59]. These two pathways have been implicated in hormonal treatment resistance both in breast [60, 61] and prostate [27, 62] cancers and are significantly enriched pathways in our datasets. The crosstalk between growth factor receptor pathways and ER signalling that activates downstream PI3K-Akt and in turn affects proliferation and survival exists in breast cancer [63]. Likewise, in PCa, ERα drives proliferation via MAPK and PI3K signalling in PTEN-null tumours [64]. As for AR, there is evidence of crosstalk with the PI3K-Akt pathway in both androgen dependent and androgen independent PCa cells [65, 66]. In addition, the signalling pathway through ERK1/2- and MEK, which is significantly reduced in our cell line data and human clinical data, has been shown to regulate PGR function [67]. Besides having nuclear receptor signalling roles, PGR is also able to indirectly activate key signalling pathways, especially, MAPK and PI3K-Akt [68, 69]. These findings further strengthen the expression balance of SHR, which ultimately can synergistically enhance other proliferative/survival pathways in CRPC.
Conclusions
Mechanisms driving CRPC are highly complex and diverse, and there is evidence for genetic differences across different populations [70] and numerous oncogenes and tumour suppressors are active in CRPC bypassing AR signalling. The nuclear receptor family, in particular, the steroid hormone receptor subfamily genes such as ER, PGR and GR may be compensating for the lack of AR activity to promote cell proliferation in CRPC. These genes may therefore represent potential drug targets in CRPC [71]. Our study also indicates that altered signalling driving cell proliferation and resistance to apoptosis is found in the LNCaP-AI cells, highlighting in particular a role for MAPK and PI3K signalling in driving proliferation, and the known links between PGR and MAPK signalling further reinforce this interpretation. Thus, our model system shows a complex, multi-layered response to prolonged androgen deprivation, and, consistent with data from human tumours, suggests that the combination of reprogrammed gene regulation compensating for the loss of AR activity and altered proliferative signalling drives the androgen resistant phenotype in this model.
Availability of supporting data
Supporting data for this paper are available in the supplementary data files referenced throughout the text.
Ethics approval
This study involved cell-based experiments and previously published publicly available data. Ethics approval was not required.
Abbreviations
- ADT:
-
androgen deprivation therapy
- Akt:
-
protein kinase B
- AR:
-
androgen receptor
- cDNA:
-
complementary DNA
- CRPC:
-
castrate resistant prostate cancer
- CS-FBS:
-
charcoal-stripped foetal bovine serum
- DBD:
-
DNA-binding domain
- DHT:
-
dihydrotestosterone
- EMT:
-
epithelial-mesenchyme transition
- ER:
-
oestrogen receptor
- GR:
-
glucocorticoid receptor
- LBD:
-
ligand- binding domain
- LHRH:
-
luteinizing hormone-releasing hormone
- MAPK:
-
mitogen-activated protein kinase
- NR:
-
nuclear receptor
- PCa:
-
prostate cancer
- PET:
-
polyethylene terephthalate
- PGR:
-
progesterone receptor
- PI3K:
-
phosphoinositide 3-kinase
- PPI:
-
protein-protein interaction
- PSA:
-
prostate specific antigen
- RNA seq:
-
RNA sequencing
- SHR:
-
steroid hormone receptor
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107.
AIHW. Australia’s health 2014. Canberra: AIHW2014 Contract No; 2014. Cat. no. AUS 178.
AIHW. Cancer in Australia: an overview 2012. Canberra: AIHW2012; 2012.
Huggins C, Hodges CV. Studies on prostatic cancer - I The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1(4):293–7.
Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34–45. doi:10.1038/35094009.
Ishikura N, Kawata H, Nishimoto A, Nakamura R, Ishii N, Aoki Y. Establishment and characterization of an androgen receptor-dependent, androgen-independent human prostate cancer cell line, LNCaP-CS10. Prostate. 2010;70(5):457–66. doi:10.1002/pros.21079.
Kawata H, Ishikura N, Watanabe M, Nishimoto A, Tsunenari T, Aoki Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate. 2010;70(7):745–54. doi:10.1002/pros.21107.
Xu G, Wu J, Zhou L, Chen B, Sun Z, Zhao F, et al. Characterization of the small RNA transcriptomes of androgen dependent and independent prostate cancer cell line by deep sequencing. PLoS ONE. 2010;5(11):e15519. doi:10.1371/journal.pone.0015519.
Takayama KI, Horie-Inoue K, Suzuki T, Urano T, Ikeda K, Fujimura T, et al. TACC2 is an androgen-responsive cell cycle regulator promoting androgen-mediated and castration-resistant growth of prostate cancer. Mol Endocrinol. 2012;26(5):748–61. doi:10.1210/me.2011-1242.
Rajan P, Sudbery IM, Villasevil ME, Mui E, Fleming J, Davis M, et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol. 2013;66(1):32–9.
Thompson IM. Flare associated with LHRH-agonist therapy. Rev in urology. 2001;3 Suppl 3:S10–4.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Ang J, Fang BL, Ashman LK, Frauman AG. The migration and invasion of human prostate cancer cell lines involves CD151 expression. Oncol Rep. 2010;24(6):1593–7.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi:10.1038/nprot.2012.016.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi:10.1093/bioinformatics/btp616.
Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, Grimmond SM, et al. PINA v2.0: mining interactome modules. Nucleic Acids Res. 2012;40(Database issue):D862–5. doi:10.1093/nar/gkr967.
Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5(9). doi:10.1371/journal.pone.0012776.
Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308. doi:10.1210/er.2002-0032.
Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol. 2014;54:49–59. doi:10.1016/j.biocel.2014.06.013.
Li Y, Vongsangnak W, Chen L, Shen B. Integrative analysis reveals disease-associated genes and biomarkers for prostate cancer progression. BMC Med Genet. 2014;7 Suppl 1:S3. doi:10.1186/1755-8794-7-s1-s3.
Tang Y, Yan W, Chen J, Luo C, Kaipia A, Shen B. Identification of novel microRNA regulatory pathways associated with heterogeneous prostate cancer. BMC Syst Biol. 2013;7 Suppl 3:S6. doi:10.1186/1752-0509-7-s3-s6.
Wang Y, Chen J, Li Q, Wang H, Liu G, Jing Q, et al. Identifying novel prostate cancer associated pathways based on integrative microarray data analysis. Comput Biol Chem. 2011;35(3):151–8. doi:10.1016/j.compbiolchem.2011.04.003.
Chen J, Wang Y, Shen B, Zhang D. Molecular signature of cancer at gene level or pathway level? Case studies of colorectal cancer and prostate cancer microarray data. Comput Math Methods Med. 2013;2013:909525. doi:10.1155/2013/909525.
Jiang J, Jia P, Zhao Z, Shen B. Key regulators in prostate cancer identified by co-expression module analysis. BMC Genomics. 2014;15:1015. doi:10.1186/1471-2164-15-1015.
Madhamshettiwar P, Maetschke S, Davis M, Reverter A, Ragan M. Gene regulatory network inference: evaluation and application to ovarian cancer allows the prioritization of drug targets. Genome Med. 2012;4(5):41.
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–49. doi:10.1038/nrc2694.
Lescarbeau RM, Kaplan DL. Quantitative analysis of castration resistant prostate cancer progression through phosphoproteome signaling. BMC Cancer. 2014;14:325. doi:10.1186/1471-2407-14-325.
Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. 2013;19(1):43–9. doi:10.1097/PPO.0b013e318282635a.
Singh SM, Gauthier S, Labrie F. Androgen receptor antagonists (antiandrogens): structure-activity relationships. Curr Med Chem. 2000;7(2):211–47.
Zielinski AJ, Fong S, Allison J, Kawahara M, Coppe JP, Feiler H, et al. The helix-loop-helix Id-1 inhibits PSA expression in prostate cancer cells. Int J Cancer. 2010;126(10):2490–6. doi:10.1002/ijc.24811.
Alimirah F, Chen J, Xin H, Choubey D. Androgen receptor auto-regulates its expression by a negative feedback loop through upregulation of IFI16 protein. FEBS Lett. 2006;580(6):1659–64. doi:10.1016/j.febslet.2006.02.015.
Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67(3):470–9. doi:10.1016/j.eururo.2014.09.049.
Germain P, Staels B, Dacquet C, Spedding M, Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58(4):685–704. doi:10.1124/pr.58.4.2.
Beato M, Klug J. Steroid hormone receptors: an update. Hum Reprod Update. 2000;6(3):225–36.
Beato M, Chavez S, Truss M. Transcriptional regulation by steroid hormones. Steroids. 1996;61(4):240–51.
Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–22. doi:10.1016/j.cell.2013.11.012.
Grindstad T, Andersen S, Al-Saad S, Donnem T, Kiselev Y, Nordahl Melbo-Jorgensen C, et al. High progesterone receptor expression in prostate cancer is associated with clinical failure. PLoS ONE. 2015;10(2):e0116691. doi:10.1371/journal.pone.0116691.
Latil A, Bieche I, Vidaud D, Lidereau R, Berthon P, Cussenot O, et al. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001;61(5):1919–26.
Lau KM, Leav I, Ho SM. Rat estrogen receptor-alpha and -beta, and progesterone receptor mRNA expression in various prostatic lobes and microdissected normal and dysplastic epithelial tissues of the Noble rats. Endocrinology. 1998;139(1):424–7. doi:10.1210/endo.139.1.5809.
Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, et al. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am J Pathol. 2001;159(1):79–92.
Savoy RM, Ghosh PM. The changing roles of steroid nuclear receptors with prostate cancer progression. Endocr Relat Cancer. 2013;20(4):C9–11. doi:10.1530/erc-13-0193.
Megas G, Chrisofos M, Anastasiou I, Tsitlidou A, Choreftaki T, Deliveliotis C. Estrogen receptor (alpha and beta) but not androgen receptor expression is correlated with recurrence, progression and survival in post prostatectomy T3N0M0 locally advanced prostate cancer in an urban Greek population. Asian J Androl. 2015;17(1):98–105. doi:10.4103/1008-682x.136445.
Zellweger T, Sturm S, Rey S, Zlobec I, Gsponer JR, Rentsch CA, et al. Estrogen receptor beta expression and androgen receptor phosphorylation correlate with a poor clinical outcome in hormone-naive prostate cancer and are elevated in castration-resistant disease. Endocr Relat Cancer. 2013;20(3):403–13. doi:10.1530/erc-12-0402.
Yao S, Till C, Kristal AR, Goodman PJ, Hsing AW, Tangen CM, et al. Serum estrogen levels and prostate cancer risk in the prostate cancer prevention trial: a nested case–control study. Cancer Causes Control. 2011;22(8):1121–31. doi:10.1007/s10552-011-9787-7.
Song CH, Lee HJ, Park E, Lee K. The chicken ovalbumin upstream promoter-transcription factor II negatively regulates the transactivation of androgen receptor in prostate cancer cells. PLoS ONE. 2012;7(11):e49026. doi:10.1371/journal.pone.0049026.
Cooney AJ, Tsai SY, O’Malley BW, Tsai MJ. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol Cell Biol. 1992;12(9):4153–63.
Klinge CM, Silver BF, Driscoll MD, Sathya G, Bambara RA, Hilf R. Chicken ovalbumin upstream promoter-transcription factor interacts with estrogen receptor, binds to estrogen response elements and half-sites, and inhibits estrogen-induced gene expression. J Biol Chem. 1997;272(50):31465–74.
Shibata H, Nawaz Z, Tsai SY, O’Malley BW, Tsai MJ. Gene silencing by chicken ovalbumin upstream promoter-transcription factor I (COUP-TFI) is mediated by transcriptional corepressors, nuclear receptor-corepressor (N-CoR) and silencing mediator for retinoic acid receptor and thyroid hormone receptor (SMRT). Mol Endocrinol. 1997;11(6):714–24. doi:10.1210/mend.11.6.0002.
Qin J, Wu SP, Creighton CJ, Dai F, Xie X, Cheng CM, et al. COUP-TFII inhibits TGF-beta-induced growth barrier to promote prostate tumorigenesis. Nature. 2013;493(7431):236–40. doi:10.1038/nature11674.
Tsai SY, Tsai MJ. Chick ovalbumin upstream promoter-transcription factors (COUP-TFs): coming of age. Endocr Rev. 1997;18(2):229–40. doi:10.1210/edrv.18.2.0294.
Perets R, Kaplan T, Stein I, Hidas G, Tayeb S, Avraham E, et al. Genome-wide analysis of androgen receptor targets reveals COUP-TF1 as a novel player in human prostate cancer. PLoS ONE. 2012;7(10):e46467. doi:10.1371/journal.pone.0046467.
Boudot A, Le Dily F, Pakdel F. Involvement of COUP-TFs in cancer progression. Cancers. 2011;3(1):700–15. doi:10.3390/cancers3010700.
Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81(1):151–7.
Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J Clin Oncol. 2005;23(9):2078–93. doi:10.1200/jco.2005.02.047.
Jones E, Pu H, Kyprianou N. Targeting TGF-β in prostate cancer: therapeutic possibilities during tumor progression. Expert Opin Ther Targets. 2009;13(2):227–34.
Zhu ML, Partin JV, Bruckheimer EM, Strup SE, Kyprianou N. TGF-beta signaling and androgen receptor status determine apoptotic cross-talk in human prostate cancer cells. Prostate. 2008;68(3):287–95. doi:10.1002/pros.20698.
van der Poel HG. Androgen receptor and TGFbeta1/Smad signaling are mutually inhibitory in prostate cancer. Eur Urol. 2005;48(6):1051–8. doi:10.1016/j.eururo.2005.09.006.
Matuszak EA, Kyprianou N. Androgen regulation of epithelial-mesenchymal transition in prostate tumorigenesis. Expert Rev Endocrinol & Metab. 2011;6(3):469–82. doi:10.1586/eem.11.32.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzym Regul. 2006;46:249–79. doi:10.1016/j.advenzreg.2006.01.004.
Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, et al. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer. 2005;12 Suppl 1:S99–111. doi:10.1677/erc.1.01005.
Zhang Y, Su H, Rahimi M, Tochihara R, Tang C. EGFRvIII-induced estrogen-independence, tamoxifen-resistance phenotype correlates with PgR expression and modulation of apoptotic molecules in breast cancer. Int J Cancer. 2009;125(9):2021–8. doi:10.1002/ijc.24540.
Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L, et al. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2013;12(11):2342–55. doi:10.1158/1535-7163.mct-13-0032.
Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005;11(2 Pt 2):865s–70.
Takizawa I, Lawrence MG, Balanathan P, Rebello R, Pearson HB, Garg E, et al. Estrogen receptor alpha drives proliferation in PTEN-deficient prostate carcinoma by stimulating survival signaling, MYC expression and altering glucose sensitivity. Oncotarget. 2015;6(2):604–16.
Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60(24):6841–5.
Squillace RM, Miller D, Wardwell SD, Wang F, Clackson T, Rivera VM. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. Int J Oncol. 2012;41(2):425–32. doi:10.3892/ijo.2012.1487.
Trevino LS, Bingman WE, Edwards DP, Nl W. The requirement for p42/p44 MAPK activity in progesterone receptor-mediated gene regulation is target gene-specific. Steroids. 2013;78(6):542–7. doi:10.1016/j.steroids.2012.12.014.
Boonyaratanakornkit V, Bi Y, Rudd M, Edwards DP. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulating gene transcription and cell cycle progression. Steroids. 2008;73(9–10):922–8. doi:10.1016/j.steroids.2008.01.010.
Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19(2):327–39. doi:10.1210/me.2004-0306.
Jiang J, Jia P, Shen B, Zhao Z. Top associated SNPs in prostate cancer are significantly enriched in cis-expression quantitative trait loci and at transcription factor binding sites. Oncotarget. 2014;5(15):6168–77.
Chen T. Nuclear receptor drug discovery. Curr Opin Chem Biol. 2008;12(4):418–26. doi:10.1016/j.cbpa.2008.07.001.
Acknowledgements
We would like to thank Dr Gayle Philip, Dr Geoff Macintyre and Dr Daniel Hurley for their assistance with setting up and running the RNA sequencing pipelines used in this study. We particularly thank Dr Geoff Macintyre for his guidance and help with the early stages of the data analysis and reviewing this work. Computational resources were provided by the Victorian Life Sciences Computational Initiative (VLSCI). MJD is funded by a National Breast Cancer Foundation Fellowship (ECF-14-43). This work was supported by grant funding to SD and AGF from Austin Medical Research Foundation and The University of Melbourne internal grant scheme.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
We certify that there is no financial and non-financial competing interests regarding the materials discussed in the manuscript.
Authors’ contributions
SD established in vitro models and carried out molecular studies including Western Blotting, Q-PCR, immunocytochemistry and motility and invasion assays. SD was responsible for the data analysis (functional studies), data interpretation, discussion and drafting and editing the manuscript. AE carried out computational experiments and was responsible for the data analysis, analysis of human data, data interpretation, discussion and drafting and editing the manuscript. EJC participated in the design of the study, interpretation of the results, and critical revision of the manuscript. MJD supervised AE, designed the computational analysis in this study, performed computational analyses and data interpretation, and participated in drafting and critical revision of the manuscript. AGF conceived of the study, supervised SD, participated in the design and coordination of this study and critically revised the manuscript. All authors read and approved the final manuscript.
Sujitra Detchokul and Aparna Elangovan are Co first authors of this work
Melissa J Davis and Albert G Frauman are Co-senior authors
Additional file
Additional file 1:
Table S1. Top 15 pathways for the human differentially expressed genes. Table S2. Top 15 pathways for the cell line deferentially expressed genes. Figure S1. Differencially expressed cell line genes overlaid on KEGG hsa04010 MAPK signaling pathway. Figure S2. Differentially expressed cell line genes overlaid on KEGG hsa04151 PI3K-Akt signaling pathway-Homo sapiens (human). Figure S3. Differentially expressed human tumour genes overlaid on KEGG hsa04010 MAPK signaling pathway. Figure S4. Differentially expressed human tumour genes overlaid on KEGG hsa04151 PI3K-Akt signaling pathway-Homo sapiens (human). (PDF 460 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Detchokul, S., Elangovan, A., Crampin, E.J. et al. Network analysis of an in vitro model of androgen-resistance in prostate cancer. BMC Cancer 15, 883 (2015). https://doi.org/10.1186/s12885-015-1884-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1884-7