
Abstract
Background
Selective BRAF inhibitors, vemurafenib and dabrafenib, and the MEK inhibitor, trametinib, have been approved for treatment of metastatic melanomas with a BRAF p.V600E mutation. The clinical significance of non-codon 600 mutations remains unclear, in part, due to variation of kinase activity for different mutants.
Methods
In this study, we categorized BRAF mutations according to the reported mutant kinase activity. A total of 1027 lung cancer, colorectal cancer or melanoma specimens were submitted for clinical mutation detection by next generation sequencing.
Results
Non-codon 600 mutations were observed in 37 % of BRAF-mutated tumors. Of all BRAF mutants, 75 % were kinase-activated, 15 % kinase-impaired and 10 % kinase-unknown. The most common kinase-impaired mutant involves codon 594, specifically, p.D594G (c.1781A > G) and p.D594N (c.1780G > A). Lung cancers showed significantly higher incidences of kinase-impaired or kinase-unknown mutants. Kinase-impaired BRAF mutants showed a significant association with concomitant activating KRAS or NRAS mutations, but not PIK3CA mutations, supporting the reported interaction of these mutations.
Conclusions
BRAF mutants with impaired or unknown kinase activity as well as concomitant kinase-impaired BRAF mutations and RAS mutations were detected in lung cancers, colorectal cancers and melanomas. Different therapeutic strategies based on the BRAF mutant kinase activity and the concomitant mutations may be worthwhile.
Similar content being viewed by others


Background
The mitogen-activated protein kinase (MAPK) or RAS/RAF/MEK/ERK signaling pathway regulates cell proliferation, differentiation and apoptosis [1]. This pathway is often dysregulated in human cancers, frequently due to activating mutations of the KRAS, NRAS, or BRAF genes. Selective BRAF inhibitors like vemurafenib and dabrafenib [2], and MEK inhibitors like trametinib have been developed to target BRAF mutant tumors [3]. Since the approval of vemurafenib by the Food and Drug Administration (FDA) of the United States in 2011 for treatment of unresectable or metastatic melanomas with a BRAF p.V600E mutation, clinical detection of the BRAF p.V600E mutation has become the standard of care for patients with metastatic melanoma in order to predict response to vemurafenib, dabrafenib and trametinib [4–7].
The BRAF gene is mutated in approximately 7 % of human cancers overall [8], specifically, 40 % to 60 % of malignant melanomas [9], 10 % to 15 % of colorectal cancers (CRCs) [10], and 1 to 5 % of non-small cell lung cancers (NSCLC) [11, 12]. While p.V600E is the most common mutation detected in many tumor types, more than 100 mutations within exons 11 and 15 of the BRAF gene have been reported in the Catalog of Somatic Mutations in Cancer (COSMIC) database, accessed on 03/10/15. The clinical significance of non-codon 600 mutations is largely unknown. In our previous retrospective study for quality assessment, next generation sequencing (NGS) demonstrated a high analytic sensitivity and a broad reportable range for clinical detection of BRAF mutations. Non-p.V600E mutants constitute a significant portion of BRAF mutations in different tumors: NSCLCs (86 %), melanomas (34 %) and CRCs (23 %) [12]. Discovering the spectrum of non-p.V600E BRAF mutations in different malignancies is a first step toward understanding their clinical significance.
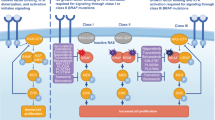
The role of BRAF mutations in the MAPK pathway is complicated not only by the multiplicity of signaling molecular components but also the variation of kinase activity for different BRAF mutants (Fig. 1). Most BRAF mutations, including the most common p.V600E (c.1799 T > A) mutation, cause upregulation of the kinase activity (kinase-activated mutants). Meanwhile, kinase-impaired BRAF mutants have also been reported [11, 13, 14]. While different assay systems have been used to determine the mutant kinase activity, most commonly, basal BRAF kinase activity was determined in vitro by measuring direct MEK phosphorylation [8, 11, 13] or measuring ERK phosphorylation in a kinase cascade assay using purified MEK and ERK proteins [15–17]. Wan et al. defined a high-activity mutant by a basal BRAF kinase activity higher than that of oncogenic RAS-activated wild-type BRAF, an intermediate-activity mutant by a basal BRAF kinase activity between those of wild-type BRAF and oncogenic RAS-activated wild-type BRAF, and a kinase-impaired mutant by a basal BRAF kinase activity lower than that of wild-type BRAF [13]. Demonstration of increased phosphorylation of BRAF and MEK proteins in patient’s tumor cell lysates [18] or inhibition of mutant-induced MEK and/or ERK phosphorylation by BRAF inhibitors in cell culture systems [19, 20] was also used to define kinase-activated mutants. Kinase-impaired mutants were further grouped into reduced-activity mutants, which could still induce MEK and ERK phosphorylation via activation of CRAF in cell culture system (Fig. 1), and silent-activity (or dead) mutant which could not [13, 16, 21]. In the presence of oncogenic RAS, however, the silent-activity mutants could induce MEK and ERK phosphorylation via activation of CRAF (Fig. 1) [22]. Since reduced-activity mutants could still activate MEK/ERK via CRAF [11, 13, 14], demonstration of mutant-induced MEK or ERK phosphorylation in cell culture systems without evidence of inhibition of mutant-induced MEK or ERK phosphorylation by BRAF inhibitors was not sufficient to define a kinase-activated mutant. Kinase-activated mutants and kinase-impaired mutants promote MEK/ERK activation and tumor progression through different mechanisms. Categorization of BRAF mutations according to their kinase activity and the presence of absence of concomitant KRAS or NRAS mutations may shed light on different therapeutic strategies to treat BRAF-mutated tumors.

Activation of MAPK pathway by kinase-activated, kinase-impaired and kinase-silent BRAF mutants through different mechanisms. Approved or potential targeted therapies are selected based on kinase activity of the BRAF mutants and concomitant mutations of the BRAF and RAS (KRAS or NRAS) genes. SBI: selective BRAF inhibitors; MEKI: MEK inhibitors; CI: CRAF inhibitors
Methods
Materials
The Johns Hopkins Medicine institutional review board (IRB) granted approval to this study with waiver of consent. A total of 1027 formalin-fixed paraffin-embedded (FFPE) neoplastic specimens with a diagnosis of lung cancer, colorectal cancer or melanoma were submitted to a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory for mutation detection using a NGS platform between April 2013 and September 2014. NGS failed in 36 (3.5 %) specimens. Fifty-two paired specimens and 3 specimens from one patient showed the same mutation patterns and were counted as one tumor per pair/triad for analysis of the prevalence and spectrum of the BRAF mutations. NGS data were available for clinical reporting in 510 lung cancers, 275 CRCs and 152 melanomas. The age of patients ranged from 33–90 (median: 65) for lung cancer specimens, 25–90 (median: 57) for CRC specimens, and 20–94 (median: 61) for melanoma specimens. The proportion of female patients was 55 % for lung cancer specimens, 45 % for CRC specimens, and 36 % for melanoma specimens. Metastatic tumors accounted for 44 % of lung cancer specimens, 28 % of CRC specimens, and 55 % of melanoma specimens. Fifteen melanoma specimens have been tested for a negative p.V600E mutation using pyrosequencing before NGS analysis. Tissue blocks with adequate tumor cellularity were selected by pathologists who made the diagnosis. One Hematoxylin & eosin (H&E) slide followed by 5–10 unstained slides and one additional H&E slide were prepared with PCR precaution. The H&E slides were examined and marked by the pathologist for subsequent macro-dissection of the FFPE neoplastic tissues from 3–10 unstained slides of 5- or 10-micron thick sections. DNA was isolated from the area(s) designated by pathologists using the Pinpoint DNA Isolation System (Zymo Research, Irvine, CA), followed by further purification via the QIAamp DNA Mini Kit (Qiagen, Valencia, CA) [23]. Tumor cellularity was also retrospectively reviewed by two molecular pathologists (GZ and MTL) as 5 quintiles (1–20 %, 21–40 %, 41–60 %, 61–80 % and 81–100 %). In the presence of discrepancy, the mean value was applied.
Next generation sequencing (NGS)
NGS was conducted using AmpliSeq Cancer Hotspot Panel (v2) for targeted multi-gene amplification as described previously [24, 25]. Briefly, we used Ion AmpliSeq Library Kit 2.0 for library preparation, Ion OneTouch 200 Template Kit v2 DL and Ion OneTouch Instrument for emulsion PCR and template preparation, and Ion PGM 200 Sequencing Kit with Ion 318 Chip and Personal Genome Machine (PGM) as the sequencing platform (Life Technologies, Carlsbad, California), all per manufacturers’ protocol. The DNA input for targeted multi-gene PCR was up to 30 ng measured by Qubit 20 Fluorometer (Life Technologies). Up to 8 specimens were barcoded using Ion Xpress Barcode Adapters (Life Technologies) for each Ion 318 chip. One to three controls (non-template control, a normal peripheral blood control from a male, and/or positive control specimens.) were included in each run. The positive control specimens were prepared from mixture of several cell lines to include mutations in the AKT, BRAF, EGFR, ERBB2, KIT, KRAS, NRAS and/or PIK3CA genes.
Sequencing data of the targeted genes were analyzed using Torrent Suite (Life Technologies). Lung cancer specimens were tested for AKT, BRAF, EGFR, ERBB2, KRAS, NRAS and PIK3CA genes (lung cancer panel), CRC specimens were tested for BRAF, KRAS, NRAS and PIK3CA genes (CRC panel), and melanoma specimens were tested for BRAF, KIT, NRAS and PIK3CA genes (melanoma panel). KRAS mutations were also analyzed for the melanoma specimens. The reference mRNA sequence was NM_005163 for AKT, NM_004333 for BRAF, NM_033360 for KRAS, NM_002524 for NRAS, and NM_006218 for PIK3CA. Mutations were identified and annotated through both Torrent Variant Caller and direct visual inspection of the binary sequence alignment/map (BAM) file on the Broad Institute’s Integrative Genomics Viewer (IGV) (http://www.broadinstitute.org/igv/). IGV was also used to determine the coverage of each specific exon and to confirm the number of reads of the variants. Novel mutations not reported in the database of COSMIC were confirmed by Sanger sequencing or pyrosequencing as described previously [12]. During our validation of this NGS assay, a cutoff of background noise at 2 % was chosen for single nucleotide variations according to a study of 16 non-neoplastic FFPE tissues [24]. With sufficient DNA input, the limit of detection is dictated by the depth of coverage (or number of sequencing reads). Approximately 150 and 500 reads is needed to detect a heterozygous mutation at a 99 % confidence in a specimen with 20 % and 10 % tumor cellularity, respectively. During the period between April 2013 and September 2014, the coverage of exon 11 and exon 15 of the BRAF gene was 1705 ± 1368 and 2182 ± 1540 reads (mean ± standard deviation), respectively.
Single Nucleotide Polymorphism (SNP) array
SNP array analysis was performed as previously described [26]. Briefly, DNA samples extracted from FFPE tissues (optimally 200 ng) were treated with the Infinium HD FFPE NDA restore kit before running on the Illumina Infinium II SNP array (HumanCytoSNP-12 v2.1 DNA Analysis BeadChip, Illumina Inc., San Diego, CA) according to manufacturer’s standard protocol. The B allele frequency and Log R ratio data were analyzed using Illumina KaryoStudio software version 2.0 and CNV (copy number variation) partition V2.4.4.0.
Reported mutant kinase activity to categorize BRAF mutations observed in the clinical specimens
The majority of mutations are predicted to cause elevation of the kinase activity (Table 1). The degree of elevation varied [13]. Mutations at codon 600 showed several hundred fold elevation of kinase activity while others showed less than 100 fold elevation. Impaired-kinase mutants involving codons 466, 469, 472, 483, 594, 596, 599 and 602 have been reported. The highly conserved aspartic acid residue encoded by codon 594 is a part of the DFG motif that plays an important role in chelating magnesium and stabilizing ATP binding [22]. Mutations at codon 594 of the BRAF genes lead to a severely reduced or silent/dead kinase with no direct or indirect activity on the downstream MAPK pathway in the absence of oncogenic RAS [13, 16, 22].
Mutations at the same codon may cause activated or impaired kinase activity depending on the specific mutation. Replacement of the conserved phosphorylation sites at codon 599 and 602 by a non-polar amino acid (such as p.T599A and p.S602A) results in complete abortion of the kinase activity while replacement by an acidic amino acid (such as p.T599E and p.S602D) leads to RAS-independent BRAF activation [17]. A discrepancy has been reported when the codon 599 residue was replaced by a bulky non-polar amino acid, isoleucine (p.T599I mutant) [13, 16]. The p.T599I mutant was categorized as an intermediate-activity mutant by direct measurement of MEK phosphorylation using ATP at a physiological concentration, but showed a slightly deceased basal kinase activity (0.84 fold) by measuring ERK phosphorylation using BRAF kinase cascade assay with ATP at a sub-physiological concentration. Another example occurs at codon 469 of the P loop which is wedged against codon 597 of the activation segment. Replacement by alanine (p.G469A) showed a 200-fold increase of basal kinase activity compared to replacement by bulky glutamic acid (p.G469E) [13]. While p.G469E was categorized as an intermediate-activity mutant, it has the lowest kinase activity within this category (1.8 fold increase) [13]. In contrast, Smalley et al. demonstrated reduced kinase activity of p.G469E mutation [21]. In the following analysis of clinical specimens, p.T599I and p.G469E were therefore grouped into the kinase-unknown category.
Statistics
Correlation between BRAF mutant allele frequency and KRAS or NRAS mutant allele frequency was examined by Spearman’s rank correlation coefficient (denoted as r) using the GraphPad Prism software (GraphPad Software, ver5, La Jolla, CA).
Results
Clinical detection of BRAF mutations in different tumors according to kinase activity
BRAF mutations were detected in 33 of 510 (6.5 %) NSCLCs, 34 of 275 (12 %) CRCs, and 67 of 152 (44 %) melanomas, including a melanoma specimen with both p.V600E (c.1799 T > A) mutation and p.S605I (c.1814G > T) occurring in the same allele (Table 2). The coverage of a BRAF mutation was 585 ± 548 reads (mean ± standard deviation). As expected, the most common residue involved by the BRAF mutation was codon 600 (86/135, 64 %), followed by codon 594 (15/135, 11 %). Non-codon 600 mutations, p.S467L (c.1400C > T) and p.G594N (c.1780G > A), were detected in 2 of 15 melanomas with prior negative pyrosequencing for codon 600. There was a significant higher fraction of non-codon 600 mutation in NSCLCs (26/33, 79 %) than those in CRCs (7/34, 21 %, P < 0.001) and melanomas (16/68, 24 %, P < 0.001).
Kinases activity is predicted to be elevated in 101 of 135 (75 %) BRAF mutations, impaired in 20 (15 %) and unknown in 14 (10 %). Unique mutations detected in only one tumor were observed in 4 of 10 unique kinase-activated mutants, 4 of 8 kinase-impaired mutants and 10 of 12 kinase-unknown mutants. The exceptions for the kinase-unknown mutant were p.G469E (c.1406G > A) and p.G469V (c.1406G > T). The most common residue involved by the kinase-impaired mutant was codon 594 (15 of 20 or 75 % of kinase-impaired mutants), specifically, p.D594G (c.1781A > G) in 8 tumors and p.D594N (c.1780G > A) in 5 tumors. Mutations involving codon 594 were observed in 7 (1.4 %) of 510 lung cancers, 4 (1.5 %) of 275 CRCs sand 4 (2.6 %) of 152 melanomas. Other kinase-impaired mutants included p. G466R (c.1396G > A), p.G466V (c.1397G > T), p.Y472C (c.1415A > G) and p.G596R (c.1786G > C). Six of 14 kinase-unknown mutants were seen in codon 469. Among those tumors with a BRAF mutation, NSCLCs showed a significant lower incidence of kinase-activated mutants (45 %) as compared to CRCs (85 %) and melanomas (84 %) and a higher incidence of kinase-impaired mutants or kinase-unknown mutants (Fig. 2). BRAF mutations with unknown kinase activity were not seen in CRC specimens.

Distribution of kinase-activated, kinase-impaired and kinase-unknown BRAF mutants. Non-small cell lung cancers (NSCLCs) showed higher incidences of kinase-impaired and kinase-unknown BRAF mutants as compared to colorectal cancers (CRCs) and melanomas
Concomitant mutations of MAPK pathway
No concomitant BRAF and EGFR or ERBB2 mutation was observed in NSCLC specimens. The frequency of concomitant KRAS or NRAS mutations found in a BRAF-mutated tumor was 4 of 33 (12 %) in NSCLCs, 3 of 34 (8.8 %) in CRCs, or 3 of 67 (4.5 %) in melanomas (Table 3). An NSCLC specimen showed an additional KRAS p.V8I (c.22G > A) mutation in the same allele of p.G13D (c.38G > A) mutation (case P10) and a melanoma specimen showed an addition PIK3CA p.P75S (c.169C > T) mutation (case P8). All except KRAS p.G15S (c.43G > A) in case P2 were activating KRAS or NRAS mutations at codon 12, 13, 59 or 61. Concomitant BRAF and activating RAS mutations were observed in 0 of 86 specimens with codon 600 mutations and in 9 of 49 specimens (18 %) with non-codon 600 mutations (Fig. 3). Concomitant BRAF and activating RAS mutations were observed in 2 of 101 (2.0 %) kinase-activated mutants, 3 of 20 (15 %) kinase-impaired mutants, and 4 of 14 (29 %) kinase-unknown mutants (Fig. 3).

Incidence of concomitant BRAF and activated RAS (KRAS or NRAS) mutations. Higher incidences of concomitant BRAF and activated RAS mutations are seen in non-codon 600 BRAF-mutated tumors (left) and in BRAF-mutated tumors with impaired or unknown kinase activity (right)
BRAF mutant allele frequencies were highly concordant with the KRAS and NRAS mutant allele frequencies (Fig. 4), suggesting that concomitant mutations are present in the same tumor population. A discrepancy was observed in cases P1 with a higher KRAS mutant allele frequency (49 % vs. 30 %), case 2 with a much lower KRAS p.G15S (c.43G > A) allele frequency (8.4 % vs. 29 %), and case P6 with a much lower BRAF p.D594N (c.1780G > A) allele frequency (5.7 % vs. 24 %). Pyrosequencing was performed in DNA specimens isolated from 3 subareas of case P1. The KRAS/ BRAF mutant allele ratio was 2.04 (49 % vs. 24 %), 1.72 (50 % vs. 29 %) and 1.59 (53 % and 34 %), respectively. SNP array analysis of case P1 revealed gain of chromosome 12p containing the KRAS gene. These results indicate that concomitant KRAS and BRAF mutations are present within the same tumor cells with amplification of the KRAS mutant allele. In case P2, the BRAF p.V600E (c.1799 T > A) mutant allele frequency (29 %) was consistent with the estimated tumor cellularity (41–60 %), suggesting KRAS p.G15S (c.43G > A) was present in a subpopulation of tumor. This was confirmed by the presence of BRAF p.V600E (c.1799 T > A) mutation in all 4 subareas, but KRAS p.G15S (c.43G > A) mutation (16 % vs. 34 % of BRAF p.V600E) in only one of 4 subareas. SNP array showed no aneuploidy of both chromosomes 12 and 7 containing KRAS gene and BRAF gene, respectively. Similarly in case P6, the BRAF p.D594N (c.1780G > A) mutant was also likely present in a subpopulation of tumors.

Correlation of mutant allele frequencies in tumors with concomitant BRAF and activating RAS mutations. The lines labeled with 80 % and 120 % indicate the boundary of events with the BRAF/RAS mutant allele ratio between 80 % and 120 %. r: Spearman’s rank correlation coefficient
The presence of 40 % BRAF and KRAS mutations in the context of 71–90 % estimated tumor cellularity in case P4 and the presence of 31 % BRAF mutation and 30 % KRAS mutation in the context of 51–70 % estimated tumor cellularity in case P5 suggest that the kinase-impaired BRAF mutation and the activating KRAS mutation were present in the all the tumor cells. KRAS/BRAF mutant allele ratio was consistently 1:1 (1.05, 1.06 and 1.00 by pyrosequencing) in 3 subareas re-isolated from cases P5, further supporting the presence of concomitant mutations in the same tumor cell population instead of different tumor subpopulations.
Concomitant mutations of mTOR pathway
Concomitant BRAF and AKT mutations were observed in 2 lung adenocarcinomas (Table 4), both of which had a BRAF p.V600E (c.1799 T > A) mutation. BRAF mutations accompanied by a PIK3CA mutation were observed in 2 of 33 (6.1 %) BRAF-mutated lung cancers, 4 of 34 (12 %) BRAF-mutated CRCs, and 2 of 67 (3.0 %) BRAF-mutated melanomas (Table 4). Concomitant BRAF and PIK3CA mutations were observed in 4 of 86 specimens (4.7 %) with a codon 600 mutation and in 4 of 49 specimens (8.2 %) with a non-codon 600 mutation (P = 0.46 by Fisher exact test). Concomitant PIK3CA mutations were observed in 4 of 101 (4.0 %) elevated-activity BRAF mutants, 1 of 20 (5.0 %) reduced-activity mutants, and 3 of 14 (21 %) unknown-activity mutants.
Discussion
BRAF mutations show diverse functional consequences and varied response to BRAF inhibitors. In this study, we categorized BRAF mutations detected in NSCLCs, CRCs and melanomas into kinase-activated mutants (75 %), kinase-impaired mutants (15 %) and kinase-unknown mutants (10 %) according to the functional studies reported in the literature. NSCLCs showed a significantly lower incidence of kinase-activated mutants than those of CRCs and melanomas. Mutations at codon 594 accounted for 11 % of BRAF mutations and were the most common kinase-impaired ones. We also demonstrated that concomitant KRAS or NRAS mutations, but not PIK3CA mutations, more likely occur with the kinase-impaired BRAF mutants than the kinase-activated ones.
While some BRAF mutations could be passenger mutations, especially those with impaired or unknown kinase activity, most BRAF mutations with elevated kinase activity are likely involved in oncogenesis and thus could be targetable. Choices of the inhibitors or inhibitor combinations are at least partly made based on BRAF mutation status. In our study, p.V600E (c.1799 T > A) was the most common BRAF mutation, occurring in 27 of 34 (79 %) CRCs, 44 of 68 (65 %) melanomas and 7 of 33 (21 %) NSCLCs. Non-p.V600E codon 600 mutations were seen in 8 of 68 (12 %) BRAF-mutated melanomas. The selective inhibitors of BRAF codon 600 mutants, vemurafenib and dabrafenib, have been shown to improve progression-free and overall survival in metastatic melanoma patients, with either the p.V600E mutation or non-p.V600E mutations at codon 600, such as p.V600K and p.V600R [4, 27–29]. Responding to vemurafenib or dabrafenib has been observed in few NSCLC patients with a BRAF p.V600E mutation [30–33], although the exact benefit of selective BRAF inhibitors for lung cancer patients is currently still under investigation [34]. Whether BRAF inhibitors benefit patients with a kinase-activated BRAF mutation located outside codon 600 remains less clear, although a partial response to vemurafenib has been reported in a melanoma patient with a p.L597R mutation [20]. Responsiveness to MEK inhibitors (TAK-733 and trametinib) in melanoma patients with a codon 597 mutations or p.K601E mutation suggests that future clinical trials of MEK inhibitors in patients with kinase-activated non-codon 600 mutations should be considered [19, 35, 36].
BRAF mutations with reduced kinase activity are unlikely responsive to BRAF inhibitors. These mutants, however, can still drive MAPK pathway through activation of CRAF/MEK/ERK cascade (Fig. 1) [13, 14]. In vitro studies of kinase-reduced mutants such as p.G466V, p.G466E and p.G596R, have also shown that activation of the CRAF/MEK/ERK cascade can be inhibited by MEK inhibitors or sorafenib, an inhibitor for multiple kinase including CRAF. In a previous clinical trial of dasatinib, a tyrosine kinase inhibitor, for metastatic non-small cell lung cancer, a patient with BRAF p.Y472C mutation remained 4-year disease-free after treatment [11]. In vitro studies confirmed the activation of CRAF/MEK/ERK cascade by the kinase-impaired p.Y472C mutant and demonstrated dasatinib-induced senescence and apoptosis in lung cancer cells expressing kinase-impaired p.G466V mutant, but not in cell lines with kinase-activated BRAF mutants.
Mutations with silent/dead kinase activity are unlikely responsive to either BRAF or MEK inhibitors. Mutations with silent/dead kinase activity have been reported in p.D594V, p.D594A and p.K483M mutations of the BRAF gene [13, 16, 22]. Codon 483 encodes the catalytic lysine and the aspartic acid at codon 594 is part of the DFG motif that plays an important role in chelating magnesium and stabilizing ATP binding [22]. Mutations at codon 483 or 594 genes lead to silence of the kinase activity with no direct or indirect activation on the downstream MAPK pathway [13, 16, 22]. Mutations at codon 594, however, have been associated with a higher incidence of co-existent RAS mutation (4 in 34, 12 %) as compared to p.V600E and presumably may also cooperate with mutations within the upstream of RAS or inter-connected pathways [22]. In this study, mutations at codon 594 constituted the second most common BRAF mutations in NSCLCs (7/33, 21 %), CRCs (4/34, 12 %) and melanomas (4/68, 5.9 %). Concomitant activating RAS mutations were observed in 2 of 15 (13 %) codon 594 mutations, but none of 86 codon 600 mutations. In the presence of oncogenic RAS proteins, kinase-silent BRAF forms a complex with CRAF and lead to hyperactivation of the CRAF/MEK/ERK cascade (Fig. 1) [22]. These preclinical studies suggested that MEK inhibitors or CRAF inhibitors may benefit patients with concomitant kinase-silent BRAF mutation and activating RAS mutation.
In contrast to the absence of concomitant BRAF p.V600E mutation and activating RAS mutation, 3 of 7 (43 %) NSCLCs and 3 of 27 (11 %) CRCs with a p.V600E (c.1799 T > A) showed a concomitant mutation in the AKT or PIK3CA genes of the mTOR pathway. In the step-wise genetic alteration model associated with colorectal tumorigenesis, PIK3CA mutations occur after KRAS or BRAF mutations and, in cooperation with other mutations, drive clonal evolution from large adenoma to invasive adenocarcinoma [37]. Thus it is not surprising to see concomitant PIK3CA mutations in 11 % of CRCs with a BRAF p.V600E (c.1799 T > A) mutation, similar to a 16 % of CRCs with an activating KRAS mutation (data not shown) in this cohort. Activating PIK3CA p.H1047R mutation has also been shown to cooperate with BRAF p.V600E mutation to promote progression of benign lung tumors to lung cancers [38]. Mutations in the AKT and PIK3CA genes, however, are uncommon in NSCLCs. PIK3CA mutations have been detected in approximately 2 % of lung adenocarcinomas [39]. The AKT p.E17K mutation was seen in only 2 of 509 NSCLCs [40–43]. In this study, the AKT p.E17K mutation was only detected in two lung adenocarcinomas with a BRAF p.V600E (c.1799 T > A) mutation, suggesting the cooperation between the MAPK and mTOR pathways, similar to that between the KRAS, NRAS or BRAF mutation and PIK3CA mutation.
In general, initiating driving mutations within the same pathway are mutually exclusive. In the setting of clinical diagnosis, caution has to be taken for interpretations of “double initiating mutations” within the same pathway. Mutations in the KRAS, NRAS and PIK3CA genes have been the mechanisms for both innate and acquired resistance to targeted therapeutics with kinase inhibitors or anti-EGFR antibodies [34]. In this study, none of patients with concomitant mutations received kinase inhibitors or anti-EGFR antibodies. In the presence of concomitant PIK3CA or AKT mutations, a combination of BRAF inhibitors or MEK inhibitors with mTOR pathway inhibitors may be more effective [44]. Correlation of the mutant allele frequencies with the estimated tumor frequency may be applied to elucidate if concomitant mutations are present in the same tumor population or only in a subpopulation. The consistency of mutant allele ratios, when testing different random subareas, would further support that concomitant mutations are present in the same population.
Conclusion
In this study, we categorized BRAF mutations according to the reported kinase activity and showed that concomitant KRAS or NRAS mutations more likely occur with the kinase-impaired BRAF mutants than the kinase-activated BRAF mutants. Different therapeutic strategies should be developed based on BRAF mutant kinase activity and the concomitant mutations.
Abbreviations
- BRAF:
-
v-raf murine sarcoma viral oncogene homologe B1
- COSMIC:
-
Catalog of Somatic Mutations in Cancer
- CRAF:
-
v-raf-1 murine leukemia viral oncogene homolog 1
- CRC:
-
Colorectal cancer
- EGFR:
-
Epidermal growth factor receptor
- ERBB2:
-
v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
- ERK:
-
Extracellular-signal-regulated kinase
- FFPE:
-
Formalin-fixed paraffin-embedded
- IGV:
-
Integrative genomics viewer
- KRAS:
-
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Mitogen-activated protein kinase kinase
- mTOR:
-
Mammalian target of rapamycin
- NGS:
-
Next generation sequencing
- NRAS:
-
Neuroblastoma RAS viral oncogene homolog
- NSCLC:
-
Non-small cell lung cancer
- PIK3CA:
-
Phosphoinositide-3-kinase-catalytic-alpha
References
Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, McCormick F. Ras signaling and therapies. Adv Cancer Res. 2009;102:1–17.
Lyle M, Long GV. The role of systemic therapies in the management of melanoma brain metastases. Curr Opin Oncol. 2014;26(2):222–9.
Grimaldi AM, Simeone E, Ascierto PA. The role of MEK inhibitors in the treatment of metastatic melanoma. Curr Opin Oncol. 2014;26(2):196–203.
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):934.
Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5(11):875–85.
Sen B, Peng S, Tang X, Erickson HS, Galindo H, Mazumdar T, et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med. 2012;4(136):136ra70.
Carter J, Tseng LH, Zheng G, Dudley J, Illei P, Gocke CD, et al. Non-p.V600E BRAF mutations are common using a more sensitive and broad detection tool. Am J Clin Pathol. 2015;144(4):620–8.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67.
Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20(6):963–9.
Houben R, Becker JC, Kappel A, Terheyden P, Brocker EB, Goetz R, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3(1):6.
Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63(23):8132–7.
Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000;19(20):5429–39.
Santarpia L, Sherman SI, Marabotti A, Clayman GL, El-Naggar AK. Detection and molecular characterization of a novel BRAF activated domain mutation in follicular variant of papillary thyroid carcinoma. Hum Pathol. 2009;40(6):827–33.
Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P, et al. BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012;2(9):791–7.
Bahadoran P, Allegra M, Le Duff F, Long-Mira E, Hofman P, Giacchero D, et al. Major clinical response to a BRAF inhibitor in a patient with a BRAF L597R-mutated melanoma. J Clin Oncol. 2013;31(19):e324–6.
Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28(1):85–94.
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21.
Lin MT, Tseng LH, Rich RG, Hafez MJ, Harada S, Murphy KM, et al. Delta-PCR, A Simple Method to Detect Translocations and Insertion/Deletion Mutations. J Mol Diagn. 2011;13(1):85–92.
Lin MT, Mosier SL, Thiess M, Beierl KF, Debeljak M, Tseng LH, et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am J Clin Pathol. 2014;141(6):856–66.
Haley L, Tseng LH, Zheng G, Dudley J, Anderson DA, Azad NS, et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol. 2015;28(10):1390–9.
Dudley JC, Gurda GT, Tseng LH, Anderson DA, Chen G, Taube JM, et al. Tumor cellularity as a quality assurance measure for accurate clinical detection of BRAF mutations in melanoma. Mol Diagn Ther. 2014;18(4):409–18.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF, Long GV. BRAF inhibitor activity in V600R metastatic melanoma. Eur J Cancer. 2013;49(5):1073–9.
McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32.
Gautschi O, Pauli C, Strobel K, Hirschmann A, Printzen G, Aebi S, et al. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol. 2012;7(10):e23–4.
Peters S, Michielin O, Zimmermann S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J Clin Oncol. 2013;31(20):e341–4.
Rudin CM, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8(5):e41–2.
Robinson SD, O’Shaughnessy JA, Cowey CL, Konduri K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer. 2014;85(2):326–30.
Morris V, Kopetz S. BRAF inhibitors in clinical oncology. F1000Prime Rep. 2013;5:11.
Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(4):482–9.
Bowyer SE, Rao AD, Lyle M, Sandhu S, Long GV, McArthur GA, et al. Activity of trametinib in K601E and L597Q BRAF mutation-positive metastatic melanoma. Melanoma Res. 2014;24(5):504–8.
Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105(11):4283–8.
Trejo CL, Green S, Marsh V, Collisson EA, Iezza G, Phillips WA, et al. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res. 2013;73(21):6448–61.
Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012;11(2):485–91.
Kim MS, Jeong EG, Yoo NJ, Lee SH. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer. 2008;98(9):1533–5.
Bleeker FE, Felicioni L, Buttitta F, Lamba S, Cardone L, Rodolfo M, et al. AKT1(E17K) in human solid tumours. Oncogene. 2008;27(42):5648–50.
Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7(5):665–9.
Do H, Solomon B, Mitchell PL, Fox SB, Dobrovic A. Detection of the transforming AKT1 mutation E17K in non-small cell lung cancer by high resolution melting. BMC Res Notes. 2008;1:14.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6.
Acknowledgements
This work was supported by National Cancer Institute, USA; grant number: 1UM1CA186691-01.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GZ, LHT and MTL carried out the study design and drafted the manuscript, GC and PI participated in the data analysis, LH carried out the pyroassay. CDG and JRE also participated in the design of the study. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, G., Tseng, LH., Chen, G. et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer 15, 779 (2015). https://doi.org/10.1186/s12885-015-1811-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1811-y