
Abstract
Background
PRDM5 is an epigenetic regulator that has been recognized as an important tumour suppressor gene. Silencing of PRDM5 by promoter hypermethylation has been demonstrated in several cancer types and PRDM5 loss results in upregulation of the Wnt pathway and increased cellular proliferation. PRDM5 has not been extensively investigated in specific subtypes of colorectal cancers. We hypothesized it would be more commonly methylated and inactivated in serrated pathway colorectal cancers that are hallmarked by a BRAF V600E mutation and a methylator phenotype, compared to traditional pathway cancers that are BRAF wild type.
Methods
Cancer (214 BRAF mutant, 122 BRAF wild type) and polyp (59 serrated polyps, 40 conventional adenomas) cohorts were analysed for PRDM5 promoter methylation using MethyLight technology. PRDM5 protein expression was assessed by immunohistochemistry in cancers and polyps. Mutation of PRDM5 was analysed using cBioPortal’s publicly available database.
Results
BRAF mutant cancers had significantly more frequent PRDM5 promoter methylation than BRAF wild type cancers (77/214,36% vs 4/122,3%; p<0.0001). Serrated type polyps had a lower methylation rate than cancers but were more commonly methylated than conventional adenomas (6/59,10% vs 0/40,0%). PRDM5 methylation was associated with advanced stages of presentation (p<0.05) and the methylator phenotype (p=0.03). PRDM5 protein expression was substantially down-regulated in both BRAF mutant and wild type cancer cohorts (92/97,95% and 39/44,89%). The polyp subgroups showed less silencing than the cancers, but similar rates were found between the serrated and conventional polyp cohorts (29/59, 49%; 23/40, 58% respectively). Of 295 colorectal cancers, PRDM5 was mutated in only 6 (2%) cancers which were all BRAF wild type.
Conclusions
Serrated pathway colorectal cancers demonstrated early and progressive PRDM5 methylation with advancing disease. Interestingly, PRDM5 protein expression was substantially reduced in all polyp types and more so in cancers which also indicates early and increasing PRDM5 down-regulation with disease progression. Methylation may be contributing to gene silencing in a proportion of BRAF mutant cancers, but the large extent of absent protein expression indicates other mechanisms are also responsible for this. These data suggest that PRDM5 is a relevant tumour suppressor gene that is frequently targeted in colorectal tumourigenesis.
Similar content being viewed by others

Background
PR (PRDI-BF1 and RIZ) domain (PRDM) proteins are a family of zinc finger transcription factors whose PR domain shares homology to the SET domain that is often present in proteins with chromatin modifying activity [1]. PRDM5 is an epigenetic regulator that does not possess this specific activity itself, however its 16 zinc fingers facilitates sequence specific protein and DNA interactions with a multitude of genes including histone methyltransferases and deacetylases [2-4]. PRDM5 recruits and directs these, specifically G9A and HDAC1, towards the promoters of its target genes to cause repression via chromatin modification [3]. PRDM5 has also been found to activate genes by maintaining RNA polymerase II at its target’s promoters [5]. Loss of PRDM5 is associated with bone morphogenic and developmental defects [5,6], and infrequent mutations of PRDM5 have been found in brittle cornea syndrome and neutropenia [3,7].
Studies have shown its promoter region contains a CpG island that is epigenetically silenced by methylation in several different cancer cell lines and primary cancers including breast, liver, gastric, lung, nasopharyngeal and esophageal [2,4,8,9]. Functional studies have identified PRDM5 as a tumour suppressor gene due to its role in suppressing cell growth and proliferation [4,8], in regulation of the cell cycle at the G2/M checkpoint [2,3] and as a heat shock responsive gene [8]. Furthermore, PRDM5 has been associated with inhibition of the Wnt pathway [6], where its overexpression prevented TCF/beta-catenin dependent transcription and repressed the downstream Wnt target, CDK4 in cancer cell lines [8]. Additionally, PRDM5 loss resulted in increased adenoma burden in mice models that had a deregulated Wnt pathway background [10].
Despite several cancers identified as having frequent PRDM5 promoter methylation, only minimal rates of methylated PRDM5 has been found in an uncharacterized series of colorectal cancers [4]. In a specific subgroup of colorectal cancers, there is frequent widespread methylation of promoter regions and subsequent silencing of key tumour suppressor genes, which is termed the CpG Island Methylator Phenotype (CIMP) [11,12]. These cancers derive from serrated type precursor lesions and are hallmarked by a V600E BRAF mutation, which with the onset of CIMP are early events in this ‘serrated pathway’ of tumourigenesis [13]. Cancers that follow the serrated pathway account for approximately 15% of all colorectal cancers. Approximately half of these cancers methylate a DNA mismatch repair gene, MLH1, and develop microsatellite instability (MSI) [14,15], and the remaining half stay as microsatellite stable (MSS).
The most common form of colorectal cancer originates from a conventional adenoma. These follow a ‘traditional pathway’ in which key molecular events, such as mutations of APC and KRAS, have been previously well defined [16] and result in cancers that are BRAF wild type and microsatellite stable.
This study has investigated whether PRDM5 methylation is a target of epigenetic silencing more commonly in the serrated compared to the traditional pathway of colorectal cancer. This was examined in both cancer and precursor lesion subgroups to give an indication of when PRDM5 is downregulated in tumourigenesis. PRDM5 protein expression was also examined in cancer and polyp subgroups, and PRDM5 mutation frequency was investigated using a publicly available database.
Methods
Patient samples
A total of 214 BRAF mutant (120 BRAF mutant/MSI and 94 BRAF mutant/MSS) and 122 BRAF wild type cancers were obtained either as fresh frozen tissue after surgical excision from the Royal Brisbane and Women’s Hospital (RBWH), Brisbane, Australia as previously described [17,18], or as formalin-fixed paraffin embedded (FFPE) tissue from Envoi Specialist Pathologists, Brisbane, Australia. Written, informed consent was obtained from each patient involved in this research which was approved by the Royal Brisbane and Women’s Hospital and Bancroft Human Research Ethics Committee. Clinicopathological data of patient gender, age at diagnosis, anatomical site of cancer (with proximal termed if proximal to the splenic flexure), and cancer stage (according to the American Joint Committee on Cancer, AJCC, system) were collected where available.
Polyp cohorts consisting of 59 serrated type polyps (19 microvesicular hyperplastic polyps, MVHPs; 20 sessile serrated adenomas, SSAs; and 20 traditional serrated adenomas, TSAs) and 40 conventional polyps (20 of each tubular adenomas, TAs; and tubulovillous adenomas, TVAs) were collected as FFPE tissue from Envoi Specialist Pathologists.
DNA from fresh cancer and matched normal tissue was extracted using AllPrep DNA mini kit (Qiagen, Dusseldorf, Germany). DNA from the FFPE cancer and polyp and matched normals were extracted by the Chelex-100 method (Bio-Rad Laboratories, CA, USA).
The presence of MSI had been previously analysed for the RBWH’s cancer samples using the National Cancer Institute’s 5 marker panel [17,19]. Cancers from Envoi Pathologists were evaluated for immunohistochemical loss of mismatch repair protein expression (MLH1, PMS2, MSH6, MSH2) as a surrogate for MSI. Presence of the BRAF V600E (a1796t) mutation, p53 mutation (over exons 4–8) and KRAS mutation (over codons 2 and 3) had been previously investigated for the RBWH’s samples [17]; presence of BRAF V600E (a1796t) and KRAS (codons 2 and 3) mutations was analysed for Envoi’s samples as previously described [17,20-22].
CpG Island Methylator Phenotype (CIMP) analysis and PRDM5 Methylation-Specific PCR
Sample DNA was bisulfite modified using Epitect Fast Bisulfite Conversion kit (Qiagen, Dusseldorf, Germany). CIMP was assessed in cancer and polyp cohorts using MethyLight technology over a 5 marker panel consisting of CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1 as previously described by Weisenberger et al. [17,22-24]. Percent of methylated reference (PMR) indicates the extent of methylation of a sample in relation to a methylated reference, and a sample with a PMR of ≥10 was considered as methylated at that marker [22]. If ≥3 markers were methylated the sample was considered CIMP-high, with 1–3 markers methylated the sample was termed CIMP-low and CIMP-0 if no markers were methylated [22]. For MSP of PRDM5, the same PMR cutoff of ≥10 applied for a sample to be considered methylated, a sample with a PMR <10 was considered unmethylated. For all CIMP markers and the PRDM5 MSPs, an Alu assay was included for each sample as a measure of the success of bisulfite conversion of that sample [22]. A cycle threshold for Alu of <23 was the sample inclusion criteria [25,26]. PRDM5 is on the reverse strand and the primer and probe sequences are as follows:
-
F: 5′AAAACTAAACAAAAACGAAAACGCA; R: 5′GGTTTTAAATTCGGAGGTTCGC;
-
Probe: 5′ 6FAM-CGCGCCGAAACTAAAAATACTAACG–BHQ1.
PRDM5 and beta catenin immunohistochemistry
Tissue sections were obtained from formalin-fixed paraffin embedded (FFPE) blocks. Antigen retrieval was performed at low pH (pH6, Reveal decloaker; Biocare Medical, CA, USA) for 15mins at 105 °C. H2O2 and Sniper were used to facilitate endogenous peroxidase and protein blocks respectively. PRDM5 antibody (anti-PRDM5, LS-1982, Lifespan BioSciences, Seattle, USA) was manually applied at 1/750 dilution and left for 1 hour. MACH3 Rabbit secondary antibody probe and polymer was applied for 10 and 20 minutes respectively (Biocare Medical, CA, USA), and DAB chromagen (Biocare Medical, CA, USA) was applied for 5 minutes. Beta-catenin antibody (anti-Beta-catenin 224 M16 (14) Cell Marque, California, USA) was manually applied and left for 1.5 hrs. MACH1 Rabbit secondary antibody probe and polymer was applied for 15 and 30 minutes respectively, and DAB was applied for 8 minutes. Sections were counterstained with haematoxylin. Slides were examined by an expert gastrointestinal pathologist and scored as either positive or negative depending on presence or absence of PRDM5 staining, and presence of nuclear beta-catenin was observed and scored either positive or negative accordingly.
Statistical analysis
Significant differences between categorical data were analysed with Fisher‘s exact test or Pearson’s chi-squared test where appropriate. Significance between continuous data was analysed by a student’s t-test. P values <0.05 were considered significant.
Results
Clinical and molecular findings of cancer cohorts
Clinical and molecular differences between the BRAF mutant and BRAF wild type cohorts concurred with previous findings [17,18]. The BRAF mutant cancers had an older age of onset, a propensity to affect females, a frequent proximal tumour location, an earlier stage at presentation and a mucinous histology compared to BRAF wild type cancers (Table 1). As expected, BRAF mutant cancers were predominantly CIMP high and had a lower rate of p53 mutation compared to BRAF wild type cancers (Table 1).
The BRAF mutant cohort was comprised of 120 MSI (56.1%) and 94 MSS (43.9%) cancers (Table 1). When clinical and molecular parameters were considered within this cohort with microsatellite status considered, the differences again correlated with previous findings [17,18]. BRAF mutant/MSS cancers affected patients at a younger average age, more frequently presented at advanced stages and were less commonly proximally located than BRAF mutant/MSI cancers (Additional file 1: Table S1). Additionally, BRAF mutant/MSS cancers were not as frequently CIMP high, but were more frequently p53 mutant compared to BRAF mutant/MSI cancers (Additional file 1: Table S1).
PRDM5 methylation in cancer cohorts
PRDM5 was methylated in 77/214 (36.0%) BRAF mutant cancers compared to 4/122 (3.3%) BRAF wild type cancers (p < 0.0001). Similarly, the average percentage of methylated reference (PMR) scores which indicates the extent of methylation of a cancer relative to a methylase treated reference sample, was significantly higher in the BRAF mutant compared to the BRAF wild type cohort (27 vs 3; p < 0.0001) (Table 1). There was no significant difference in PRDM5 methylation rates within the BRAF mutant cohort when stratified for microsatellite status (Additional file 1: Table S1).
BRAF mutant cancers that had methylated PRDM5 were more likely to present at advanced stages compared to BRAF mutant cancers with unmethylated PRDM5 (AJCC stage III/IV: 29/65, 44.6% vs 31/105, 29.5%; p < 0.05) (Table 2). PRDM5 methylation correlated with CIMP high. CIMP high was strongly prevalent in the BRAF mutant/MSI cancers (at 86%), therefore this was evident in the BRAF mutant/MSS cohort (61% CIMP high rate) where CIMP high was more frequent in PRDM5 methylated compared to unmethylated cancers (27/36, 75.0% vs 27/53 50.9%; p = 0.03) (Additional file 1: Table S2).
PRDM5 protein expression in cancer cohorts
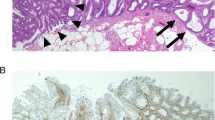
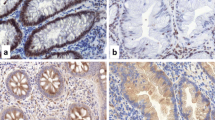
Immunohistochemical analysis of PRDM5 protein expression in adjacent normal mucosa showed it was routinely present within the crypt bases. Interestingly, there was substantial loss of protein expression in both the BRAF mutant (92/97, 94.5%) and BRAF wild type (39/44, 88.6%) cancer cohorts (Figure 1).

PRDM5 immunohistochemistry. A demonstrates the normal pattern of staining in non-neoplastic colonic mucosa. Down arrows indicate strong nuclear and cytoplasmic staining for PRDM5 in scattered cells predominantly in the crypt bases. Up arrowheads indicate incidental melanosis coli in the lamina propria. B is a representative area of a colorectal carcinoma negative for PRDM5. C is a representative area of a colorectal carcinoma positive for PRDM5, showing scattered cells with strong cytoplasmic and/or nuclear staining. (Original magnification: x200).
Of the 10 cancers with retained PRDM5 protein expression, 9 had unmethylated PRDM5. The high rate of PRDM5 protein loss compared to the rate of methylation across all cancers, and the high frequency of absent PRDM5 protein expression observed in unmethylated cancers, clearly indicates that other mechanisms besides methylation are contributing to PRDM5 protein down-regulation.
PRDM5 methylation and protein expression in polyp cohorts
Fifty-nine serrated type precursor lesions (19 MVHPs, 20 SSAs, 20 TSAs), and forty conventional type precursor lesions (20 TAs, 20 TVAs) were included in the analysis. Molecularly, the serrated polyps were significantly more methylated and BRAF mutant as expected, and all polyp subtypes had a low KRAS mutation rate (Table 3). Clinically, the TVAs and TSAs were more likely to be larger and distally located (Additional file 1: Table S3).
Methylation and protein expression of PRDM5 was analysed across all polyp subgroups to determine whether down-regulated PRDM5 was an early event in tumourigenesis and in which polyp type or pathway this was mostly occurring in.
PRDM5 was methylated in 2 of each of the three serrated polyp subtypes to give an overall methylation rate of 10% (6/59) in the serrated polyps compared to the lack of methylation in both subtypes of conventional polyps (0/40) (p = 0.08). The average PMR of methylated PRDM5 was significantly higher in all serrated polyp subtypes compared to the conventional polyps (p = 0.01) (Table 3).
PRDM5 protein expression was reduced across the serrated SSAs and TSAs, and conventional TAs and TVAs polyp subgroups at similar frequencies (an average of 59%). MVHPs which are the earliest form of serrated lesion, had a lower rate of loss (at 26%) compared to SSAs (60%) and TSAs (45%), which suggests there is a progressive down-regulation of PRDM5 with advancing disease (Table 3). Due to the greater rates of PRDM5 protein loss compared to methylation, other mechanisms are contributing to this silencing especially in the conventional polyps, as was seen in the cancer cohorts.
As expected, the rate of BRAF V600E mutation and CIMP high was significantly more common in serrated type polyps than conventional polyps (both p < 0.0001), and KRAS mutation in codons 2 and 3 was relatively low across both serrated and conventional polyp types (8% and 10% respectively) (Table 3).
PRDM5 mutation analysis
A publicly available database was searched for presence of PRDM5 mutations in colorectal cancer. cBioPortal (www.cBioPortal.org) [27] incorporates data from The Cancer Genome Atlas Network’s colorectal cancer study [28] and Seshagiri et al. 2010 [29]. Collectively there are 296 colorectal cancers with somatic mutation data that show an 8.1% BRAF V600E mutation rate. In total, 7 PRDM5 mutations were reported from 6 cancer samples (2.0% mutation rate) that were all BRAF wild type. These mutations were spread along the length of the gene and no two were similar. Due to the low rate of PRDM5 mutation found, this type of analysis was not extended to this study’s cancer or polyp cohorts.
Presence of nuclear beta-catenin in cancer and polyp subgroups
Nuclear beta-catenin as a surrogate of Wnt pathway activation was present in 39% (36/92) BRAF mutant cancers which was significantly lower than the rate observed in BRAF wild type cancers at 86% (36/42) (p < 0.0001) (Table 1). As expected, there was also a significantly reduced rate of nuclear beta-catenin in serrated compared to conventional polyps (3/59, 5% vs 26/40, 65%) (p < 0.0001) (Table 3).
Due to previous correlations of methylated PRDM5 with presence of active beta-catenin and exogenous PRDM5 causing a decrease in downstream Wnt reporter assays [8], presence of methylated PRDM5 and nuclear beta-catenin was assessed within the cancer cohorts.
There was no significant association of nuclear beta-catenin occurring in PRDM5 methylated compared to unmethylated BRAF mutant cancers (16/33; 49% vs 20/59, 34%) (p = 0.2) (Table 2). Only when stratified for MSI status, was a correlation observed with a higher rate of nuclear beta-catenin in PRDM5 methylated compared to unmethylated BRAF mutant/MSI cancers (13/21, 62% vs 12/38, 32%) (p = 0.03) (Additional file 1: Table S2). No correlation with presence of nuclear beta-catenin and methylated PRDM5 was seen in BRAF mutant/MSS cancers.
Discussion
This study investigated a large series of molecularly subtyped colorectal cancers and precursor lesions for the presence of PRDM5 methylation and protein expression. We found the PRDM5 promoter region was substantially methylated in BRAF mutant cancers of the serrated pathway whereas minimal levels of methylation were detected in the BRAF wild type cancers of the traditional pathway. This is the first study to show that a particular subgroup of colorectal cancer has a comparably high rate of PRDM5 methylation as previously found in other cancers such as lung, breast, liver and gastric cancer [2,4,8,9].
PRDM5 methylation was evident in a small proportion of serrated type polyps which indicates this may be an early event in tumourigenesis in the serrated pathway. The frequency of BRAF mutation and CIMP increased from serrated polyp to cancer as expected. The frequency of PRDM5 methylation also increased from serrated precursor lesion to BRAF mutant cancers at a similar proportion which suggests that PRDM5 methylation associates with advancing disease in cancers of the serrated pathway. Furthermore, there was an association of PRDM5 methylation being more prevalent in BRAF mutant cancers presenting at late compared to early stages which further indicates epigenetic regulation of PRDM5 may influence disease progression in the serrated pathway. This association was also seen in a previous study where PRDM5 methylation was more common in high grade breast and liver cancers [2].
The absence of PRDM5 methylation found in conventional adenomas and the low rate seen in BRAF wild type cancers indicates that PRDM5 methylation is not an important event in traditional pathway cancers. This minimal PRDM5 methylation rate in BRAF wild type cancers, at 3%, was similar to the low frequency found in the one other study that investigated primary colorectal cancers [4], and others have concluded that there is only a negligible rate of PRDM5 methylation in colorectal cancer based on cell line analysis [8]. The findings from this study highlights the importance of stratifying for molecular subtype with analysis of molecular markers involved in colorectal cancer as it is a heterogenous disease comprised of several clinically and genetically distinct subtypes. Although the KRAS mutation rate was minimal in the conventional adenoma cohorts, similarly low rates, particularly for TAs, have been previously found [20,30], and wide variations of KRAS mutation rates in adenomas have been reported [31-33].
CIMP is highly prevalent in cancers of the serrated pathway, particularly those that are microsatellite unstable. When the BRAF mutant cancers were stratified for MSI status, it was apparent that PRDM5 methylation correlated with CIMP. However, it is unlikely that PRDM5 methylation is merely a passenger event of CIMP. This is due to there being a considerable presence of PRDM5 methylation and transcript down-regulation in several non-CIMP cancer types [2,4,8,9], and there is a lack of reported PRDM5 methylation in other CIMP related cancers such as glioma. Additionally, the substantial loss of PRDM5 protein expression found in this study, suggests that potentially methylation and loss of PRDM5 is highly relevant in tumourigenesis [34].
Endogenous PRDM5 protein expression was routinely detected in normal tissue sections in this study which concurs with a previous investigation that found PRDM5 transcript expression was prevalent in several normal tissues [8]. Interestingly the vast majority of cancers in both the BRAF mutant and BRAF wild type cancer cohorts lacked PRDM5 protein expression. Absent expression was also widespread in both serrated and conventional polyps, although this rate of downregulation was less than that in the cancers. MVHPs which are the earliest form of serrated lesion and may give rise to SSAs, had the least frequency of absent PRDM5 protein expression, and overall this analysis demonstrates an early and linear progression of downregulated PRDM5 with advancing disease across all colorectal subgroups of both the serrated and traditional pathways.
This frequent loss of PRDM5 protein expression seen by immunohistochemistry is concordant with findings of a previous study that investigated expression in 18 colorectal cancers that were not molecularly subtyped [10]. However, half of this study’s normal sections had no observed endogenous protein which may indicate the inability of the antibody used to reliably detect protein within their cancer samples. Of the 10 cancers in the present study that retained PRDM5 protein expression, there was 90% concordance with these cancers being unmethylated. The one cancer that was methylated but still expressed PRDM5 protein may be in the seeding stages of methylation, and although the relatively few CpG sites covered by the methylight assay were methylated, they were not sufficient to fully silence protein expression. Additionally, the cancers that were methylated with concordant absent expression, may represent the presence of a more global methylation pattern driving protein loss in these cancers. Similar incidences of methylated gastric and esophageal cancer cell lines showing positive transcript expression has been observed [8]. This study’s methylight assay was in very close proximity to the MSP of Shu et al’s [8] in the promoter region which helps to further suggest that in some cancers, extensive methylation over the promoter is required for complete down-regulation.
Previous studies have mostly analysed PRDM5 transcript expression as a measure of the extent of silencing [2,4,8,35]. Although one reported similar findings to this current study where decreased transcript expression was observed in unmethylated gastric cancers [4], concordance was found between reduced transcript expression and methylation of nasopharyngeal cancers, and therefore silencing induced primarily by methylation was concluded [8]. This current study analysed protein expression which is a more relevant determinant of the functional endpoint state of the gene and it reflects any post translational modifications that may have taken place. Results showed a far greater rate of loss compared to methylation frequency, indicating that in colorectal cancer methylation is just one of the mechanisms responsible for this.
PRDM5 mutation events contribute to brittle cornea syndrome and neutropenia [3,7], however they have not been analysed in cancer types previously. This study utilised a publicly available database, cBioPortal [27], which incorporates data from two large series of colorectal cancers [28,29]. Overall a low rate of mutation was found and there was no identifiable mutational hotspot which indicated this mechanism is not a common cause of down-regulation. However, all mutations were present in BRAF wild type cancers which may still indicate this mechanism is of some relevance in traditional pathway cancers.
PRDM5 is located on chromosome 4q27 which is within a region commonly deleted in colorectal cancer [36,37]. Analysis of recent SNP array data, revealed loss over this locus in 33% BRAF mutant/MSS and 44% BRAF wild type cancers [38], which suggests gene deletion may also contribute to the levels of down-regulation observed. It was found that the colorectal cancer cell line, SW480, lacked PRDM5 expression due to methylation of histone H3K27 and not as a result of a methylated promoter region [4]. Therefore, histone modification events that can alter chromatin structure and result in gene suppression provide a further mechanism of PRDM5 silencing. Additionally, small and long regulatory RNAs may be acting at both the post-transcriptional and pos-translational stages to effect gene and /or protein expression [39], as well as one of the many other post-translational modifications such as acetylation and that could be taking place to affect expression.
The Wnt pathway is one of the most aberrantly upregulated pathways present in colorectal cancer [28]. Previous findings have shown that PRDM5 can interact with a variety of genes involved in inhibition of the Wnt pathway [6], PRDM5 loss results in an increased number of intestinal adenomas on an upregulated Wnt background [10], and methylated PRDM5 has been correlated with presence of active beta-catenin in cancer cell lines [8]. In this study, methylated PRDM5 associated with presence of nuclear beta-catenin in the BRAF mutant/MSI cancers (Additional file 1: Table S2). These cancers, through their heavily methylated phenotype have been found to methylate other inhibitors of the Wnt pathway such as DKK1 and AXIN2 [40,41], which indicates epigenetic regulation of the Wnt pathway may be more prevalent in the BRAF mutant/MSI compared to other CRC subtypes.
Conclusions
This is the first study that has analysed the rate of PRDM5 methylation and protein expression in a large and well characterized series of colorectal cancer and polyp subgroups.
PRDM5 methylation was found to be an early event with progressive acquisition in BRAF mutant cancers of the serrated pathway. Furthermore, PRDM5 protein levels were substantially reduced across both serrated and conventional polyp types and more so in BRAF mutant and wild type cancers. This indicates that down-regulation is initiated early in tumourigenesis and is progressive with disease advancement in both the serrated and traditional pathways. Epigenetic modification may be contributing to gene silencing in a proportion of BRAF mutant cancers and the large extent of absent protein expression indicates other mechanisms are also responsible for PRDM5 down-regulation. PRDM5 mutation was present in a small percentage of BRAF wild type cancers and this may be a cause of downregulation in this cancer subgroup. Overall, this investigation highlights PRDM5 as an important tumour suppressor gene in colorectal cancer.
Abbreviations
- AJCC:
-
American Joint Committee on Cancer
- CIMP:
-
CpG Island Methylator Phenotype
- CRC:
-
Colorectal cancer
- FFPE:
-
Formalin-fixed paraffin embedded
- MSI:
-
Microsatellite instability
- MSP:
-
Methylation-specific PCR
- MSS:
-
Microsatellite stable
- MVHP:
-
Micro-vesicular hyperplastic polyp
- PMR:
-
Percent of methylated reference
- PRDM:
-
PRDI-BF1 and RIZ domain
- SSA:
-
Sessile serrated adenoma
- TA:
-
Tubular adenoma
- TSA:
-
Traditional serrated adenoma
- TVA:
-
Tubulovillous adenoma
References
Huang S, Shao G, Liu L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem. 1998;273(26):15933–9.
Deng Q, Huang S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene. 2004;23(28):4903–10.
Duan Z, Person RE, Lee HH, Huang S, Donadieu J, Badolato R, et al. Epigenetic regulation of protein-coding and microRNA genes by the Gfi1-interacting tumor suppressor PRDM5. Mol Cell Biol. 2007;27(19):6889–902.
Watanabe Y, Toyota M, Kondo Y, Suzuki H, Imai T, Ohe-Toyota M, et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin Cancer Res. 2007;13(16):4786–94.
Galli GG, Honnens de Lichtenberg K, Carrara M, Hans W, Wuelling M, Mentz B, et al. Prdm5 regulates collagen gene transcription by association with RNA polymerase II in developing bone. PLoS Genet. 2012;8(5):e1002711.
Meani N, Pezzimenti F, Deflorian G, Mione M, Alcalay M. The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS One. 2009;4(1):e4273.
Burkitt Wright EM, Spencer HL, Daly SB, Manson FD, Zeef LA, Urquhart J, et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet. 2011;88(6):767–77.
Shu XS, Geng H, Li L, Ying J, Ma C, Wang Y, et al. The epigenetic modifier PRDM5 functions as a tumor suppressor through modulating WNT/beta-catenin signaling and is frequently silenced in multiple tumors. PLoS One. 2011;6(11):e27346.
Tan SX, Hu RC, Tan YL, Liu JJ, Liu WE. Promoter methylation-mediated downregulation of PRDM5 contributes to the development of lung squamous cell carcinoma. Tumour Biol. 2014;35(5):4509–16.
Galli GG, Multhaupt HA, Carrara M, de Lichtenberg KH, Christensen IB, Linnemann D, et al. Prdm5 suppresses Apc-driven intestinal adenomas and regulates monoacylglycerol lipase expression. Oncogene. 2014;33(25):3342–50.
Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59(10):2307–12.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96(15):8681–6.
Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138(6):2088–100.
Koinuma K, Shitoh K, Miyakura Y, Furukawa T, Yamashita Y, Ota J, et al. Mutations of BRAF are associated with extensive hMLH1 promoter methylation in sporadic colorectal carcinomas. Int J Cancer. 2004;108(2):237–42.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–11.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67.
Bond CE, Umapathy A, Ramsnes I, Greco SA, Zhen Zhao Z, Mallitt KA, et al. p53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int J Cancer. 2012;130(7):1567–76.
Bond CE, Umapathy A, Buttenshaw RL, Wockner L, Leggett BA, Whitehall VL. Chromosomal instability in BRAF mutant, microsatellite stable colorectal cancers. PLoS One. 2012;7(10):e47483.
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57.
Spring KJ, Zhao ZZ, Karamatic R, Walsh MD, Whitehall VL, Pike T, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology. 2006;131(5):1400–7.
Zhao ZZ, Nyholt DR, Le L, Martin NG, James MR, Treloar SA, et al. KRAS variation and risk of endometriosis. Mol Hum Reprod. 2006;12(11):671–6.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–93.
Weisenberger DJ, Campan M, Long TI, Kim M, Woods C, Fiala E, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33(21):6823–36.
Whitehall VL, Rickman C, Bond CE, Ramsnes I, Greco SA, Umapathy A, et al. Oncogenic PIK3CA mutations in colorectal cancers and polyps. Int J Cancer. 2012;131(4):813–20.
Burnett-Hartman AN, Newcomb PA, Potter JD, Passarelli MN, Phipps AI, Wurscher MA, et al. Genomic aberrations occurring in subsets of serrated colorectal lesions but not conventional adenomas. Cancer Res. 2013;73(9):2863–72.
Bettington M WN, Rosty C, Brown I, Clouston A, McKeone D, Pearson S, Leggett B, Whitehall V: A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Modern Pathology. 2014, in print.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–4.
Fernando WC, Miranda MS, Worthley DL, Togashi K, Watters DJ, Leggett BA, et al. The CIMP Phenotype in BRAF Mutant Serrated Polyps from a Prospective Colonoscopy Patient Cohort. Gastroenterol Res Pract. 2014;2014:374926.
Kakar S, Deng G, Cun L, Sahai V, Kim YS. CpG island methylation is frequently present in tubulovillous and villous adenomas and correlates with size, site, and villous component. Hum Pathol. 2008;39(1):30–6.
Yagi K, Takahashi H, Akagi K, Matsusaka K, Seto Y, Aburatani H, et al. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol. 2012;180(2):616–25.
Yadamsuren EA, Nagy S, Pajor L, Lacza A, Bogner B. Characteristics of advanced- and non advanced sporadic polypoid colorectal adenomas: correlation to KRAS mutations. Pathol Oncol Res. 2012;18(4):1077–84.
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55.
Cheng HY, Chen XW, Cheng L, Liu YD, Lou G. DNA methylation and carcinogenesis of PRDM5 in cervical cancer. J Cancer Res Clin Oncol. 2010;136(12):1821–5.
Malkhosyan S, Yasuda J, Soto JL, Sekiya T, Yokota J, Perucho M. Molecular karyotype (amplotype) of metastatic colorectal cancer by unbiased arbitrarily primed PCR DNA fingerprinting. Proc Natl Acad Sci U S A. 1998;95(17):10170–5.
Arribas R, Risques RA, Gonzalez-Garcia I, Masramon L, Aiza G, Ribas M, et al. Tracking recurrent quantitative genomic alterations in colorectal cancer: allelic losses in chromosome 4 correlate with tumor aggressiveness. Lab Invest. 1999;79(2):111–22.
Bond CE, Nancarrow DJ, Wockner LF, Wallace L, Montgomery GW, Leggett BA, et al. Microsatellite stable colorectal cancers stratified by the BRAF V600E mutation show distinct patterns of chromosomal instability. PLoS One. 2014;9(3):e91739.
Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108(12):2419–25.
Rawson JB, Manno M, Mrkonjic M, Daftary D, Dicks E, Buchanan DD, et al. Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis. 2011;32(5):741–7.
Koinuma K, Yamashita Y, Liu W, Hatanaka H, Kurashina K, Wada T, et al. Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene. 2006;25(1):139–46.
Acknowledgements
This study and authors were funded by a National Health and Medical Research Council grant (NHMRC: 1050455). In addition, Catherine Bond was funded by an Australian Postgraduate Award, Mark Bettington was funded by a Cancer Council Queensland scholarship, and Barbara Leggett and Vicki Whitehall received funding from Queensland Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CB assisted in the design of the study, performed experimental work, analysed data and drafted the manuscript. MB assisted with the methylight methodology, analysed immunohistochemistry investigations and generated the immunohistochemistry images. SP optimized and assisted with the immunohistochemistry investigations. DM assisted with the methylight methodology. BL helped with the design and coordination of the study. VW conceived and helped to design the study and analyse data. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Clinical and Molecular Features of Cancer Cohorts Stratified by MSI Status; Clinical and Molecular Features of BRAF mutant cohorts stratified by PRDM5 Methylation Status; Clinical data for serrated polyp and conventional adenoma cohorts.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bond, C.E., Bettington, M.L., Pearson, SA. et al. Methylation and expression of the tumour suppressor, PRDM5, in colorectal cancer and polyp subgroups. BMC Cancer 15, 20 (2015). https://doi.org/10.1186/s12885-015-1011-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-015-1011-9