
Abstract
Background
Optimisation of dopaminergic therapy may alleviate fluctuation-related pain in Parkinson’s disease (PD). Opicapone (OPC) is a third-generation, once-daily catechol-O-methyltransferase inhibitor shown to be generally well tolerated and efficacious in reducing OFF-time in two pivotal trials in patients with PD and end-of-dose motor fluctuations. The OpiCapone Effect on motor fluctuations and pAiN (OCEAN) trial aims to investigate the efficacy of OPC 50 mg in PD patients with end-of-dose motor fluctuations and associated pain, when administered as adjunctive therapy to existing treatment with levodopa/dopa decarboxylase inhibitor (DDCi).
Methods
OCEAN is a Phase IV, international, multicentre, randomised, double-blind, placebo-controlled, parallel-group, interventional trial in PD patients with end-of-dose motor fluctuations and associated pain. It consists of a 1-week screening period, 24-week double-blind treatment period and 2-week follow-up period. Eligible patients will be randomised 1:1 to OPC 50 mg or placebo once daily while continuing current treatment with levodopa/DDCi and other chronic, stable anti-PD and/or analgesic treatments. The primary efficacy endpoint is change from baseline in Domain 3 (fluctuation-related pain) of the King’s Parkinson’s disease Pain Scale (KPPS). The key secondary efficacy endpoint is change from baseline in Domain B (anxiety) of the Movement Disorder Society-sponsored Non-Motor rating Scale (MDS-NMS). Additional secondary efficacy assessments include other domains and total scores of the KPPS and MDS-NMS, the Parkinson’s Disease Questionnaire (PDQ-8), the MDS-sponsored Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Parts III and IV, Clinical and Patient’s Global Impressions of Change, and change in functional status via Hauser’s diary. Safety assessments include the incidence of treatment-emergent adverse events. The study will be conducted in approximately 140 patients from 50 clinical sites in Germany, Italy, Portugal, Spain and the United Kingdom. Recruitment started in February 2021 and the last patient is expected to complete the study by late 2022.
Discussion
The OCEAN trial will help determine whether the use of adjunctive OPC 50 mg treatment can improve fluctuation-associated pain in PD patients with end-of-dose motor fluctuations. The robust design of OCEAN will address the current lack of reliable evidence for dopaminergic-based therapy in the treatment of PD-associated pain.
Trial registration
EudraCT number 2020–001175-32; registered on 2020-08-07.
Similar content being viewed by others


Background
Although fragmented accounts of Parkinsonism date back to 2500 BC, initial descriptions of its cardinal motor signs were much more recent (as in the 1690 book, ‘Pax corporis’, by Ferenc Pápai Páriz); however, it was only in 1817 that James Parkinson medically described the motor symptoms of the disease in such detail that the condition would subsequently be named after him [1,2,3].
Although the relationship between motor symptoms and non-motor symptoms (NMS) is variable and not necessarily linear, many NMS – specifically pain – can undergo fluctuations based on ON and OFF states during long-term treatment with levodopa [4, 5]. NMS that fluctuate in parallel with motor symptoms and in relationship to plasma levodopa levels have been termed non-motor fluctuations (NMF) [6] and encompass a range of neuropsychiatric (e.g. depression, apathy, fatigue), autonomic (e.g. sweating, micturition frequency/urgency), cognitive and sensory (e.g. pain) manifestations [5, 7, 8]. NMF have been reported to occur in 60–100% of patients with motor fluctuations, and may result in greater disability and burden than motor disturbances [5, 7]. NMF are complex, their appearance not always matching that of motor fluctuations in terms of timing [4, 8], and their underlying pathogenic mechanisms are still relatively unclear; however, sizable evidence suggests that, similarly to motor fluctuations, involvement of the dopaminergic system is key, with dopamine either being directly involved or working as a modulator of serotonin, norepinephrine or acetylcholine [7, 8]. In contrast to the relatively linear progression of most motor features during the disease course of PD, some NMS increase in frequency while others improve as dopaminergic therapy is initiated [8,9,10]; NMF are among those usually responsive to dopaminergic therapy optimisation [4, 7].
Pain is one of the most frequent and burdensome NMS in PD, being a significant comorbidity in up to 85% of PD patients, and may precede motor symptoms of the disease [11,12,13,14,15,16]. The pathophysiology underlying pain in PD is complex and not completely elucidated, and its management remains a key unmet need. The types and distribution of pain experienced by patients with PD are heterogeneous (Table 1) [17,18,19,20]. Spontaneous pain may be triggered by disease-related and/or comorbid conditions, exacerbated by a lowered pain threshold that may result from dysfunctional nociceptive processing caused by specific neurodegenerative changes [12, 21, 22]. Abnormal basal ganglia function in PD modulates pain directly and indirectly via mechanisms that impact both affective and cognitive nociceptive processing [23]. Pain has been shown to be associated with sleep disruption and cardiovascular disturbances in PD, and there is an indication that pain, sleep disruption and dysautonomia may share a common pathophysiology involving non-dopaminergic pathways [16]. Several recent publications have reviewed the current treatment options for each type of pain in PD [12, 19, 24, 25].
Pain in PD is also associated with motor fluctuations [11, 26]. The role of dopamine in pain signalling is complex. Pain relief elicits rewards mediated by elevated dopamine in the nucleus accumbens, and reciprocity with higher brain regions such as the anterior cingulate cortex and dopaminergic transmission therein is necessary for the relief of pain aversiveness [27]. Meanwhile, along with serotonin and noradrenaline, dopamine may modulate pre-synaptic inhibition in the mouse spinal cord [28]. The precise mechanism underlying dopamine’s role in pain modulation is hitherto equivocal. Clinically, dopaminergic therapies have been shown to alleviate pain in PD [12, 21, 22, 29] and fluctuation-related pain in PD is believed to be partially mediated by dopamine [12, 30]. Studies that manipulate dopamine with the aim of translation to clinical therapy will be hampered by dopamine’s role in movement and reward [31]. While optimisation of dopaminergic therapy may alleviate fluctuation-related pain [12, 22, 32, 33], high-quality evidence of the benefit of dopaminergic therapies in PD-associated pain is lacking [30], with only one study coming close to providing Level 1 evidence, although failing to meet its primary endpoint at 16 weeks [34]. One reason for this is that, until recently, there were no disease-specific scales to adequately measure the heterogeneous types of pain in PD; this has now been resolved with the development and validation of the King’s Parkinson’s disease Pain Scale (KPPS) [35].
Levodopa is still the most effective symptomatic treatment for PD [36]. However, following oral administration, levodopa is extensively metabolised in the periphery by dopa decarboxylase (DDC) and catechol-O-methyltransferase (COMT), and only 1% of an oral dose of levodopa reaches the brain [37, 38]. Moreover, long-term treatment with levodopa is complicated by the development of wearing off and drug-induced dyskinesia [36, 39]. Pain increases during OFF periods and patients with dyskinesia have increased pain sensitivity [40, 41]. Treatment with levodopa has been shown to improve pain thresholds in patients with PD, unlike the dopamine agonist apomorphine [16, 42]. Inhibitors of DDC (DDCi) and COMT (COMTi) are commonly used as an adjunct to levodopa in patients with PD in order to increase levodopa bioavailability and its delivery to the brain, and thereby ameliorate wearing-off symptoms [38, 43, 44].
Opicapone (OPC) is a third-generation, once-daily COMTi [37, 38, 45, 46], which has been shown to be generally well tolerated and efficacious in reducing OFF-time in two pivotal trials in patients with PD and end-of-dose motor fluctuations (BIPARK-I and II) [47, 48]. On the basis of these trials, OPC is approved in the European Union, USA, Japan, Australia and other countries as adjunctive therapy to preparations of levodopa/DDCi in patients with PD and end-of-dose motor fluctuations [49] or OFF episodes [50]. A positive signal for OPC was observed on the Non-Motor Symptoms Scale (NMSS) miscellaneous domain, which includes pain, in both the BIPARK II trial [51] and the OPTIPARK study [52].
Given the probable dopamine-related pathophysiology of motor fluctuation-associated pain [12] and the encouraging signals detected in previous OPC studies, the OpiCapone Effect on motor fluctuations and pAiN (OCEAN) study has been designed. This trial aims to investigate the efficacy of OPC 50 mg in PD patients with end-of-dose motor fluctuations and associated pain, when administered as adjunctive therapy to existing treatment with levodopa/DDCi.
Methods/design
Study design
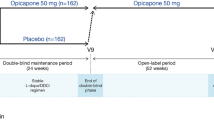
OCEAN is a Phase IV, international, multicentre, randomised, double-blind, placebo (PLC)-controlled, parallel-group, interventional trial in PD patients with end-of-dose motor fluctuations and associated pain (experienced for ≥4 weeks prior to the start of the study, with a score of ≥12 [out of 36] on Domain 3 of the KPPS at screening and baseline). It consists of a 1-week screening period, 24-week double-blind treatment period and 2-week follow-up period (Fig. 1). Following screening, at visit (V) 2 (baseline), eligible patients will be randomised 1:1 to OPC 50 mg or PLC once daily while continuing current treatment with levodopa/DDCi. Since OPC enhances the effects of levodopa, it may be necessary to reduce the patient’s levodopa/DDCi dosing within the first days or weeks of OPC treatment; therefore, the investigator may decrease the daily dose of levodopa/DDCi as needed until V4, while keeping the number of daily intakes unchanged. If necessary, dosing may be increased back to the baseline dose level. After V4, the levodopa/DDCi dose should not be changed until the end of the study. The anti-PD treatment regimen should be stable for at least 4 weeks prior to V1 (Table 2) and kept stable throughout the study (except for levodopa/DDCi during the adjustment period). No new anti-PD drugs should be started during the study.

Study design. aV2 is divided in V2a and V2b. If ON/OFF diary entries are non-compliant at V2a, the patient will be re-trained on correct use of the diary and visit V2b will be postponed for 3–4 days. If diary completion is satisfactory at V2a, V2b is performed immediately on the same day. AE, adverse event; DDCI, dopa decarboxylase inhibitor; L-dopa, levodopa; PD, Parkinson’s disease; PSV, post-study visit; V, visit
Chronic pain treatment should be stable for at least 4 weeks prior to V1 (Table 2), and no new pain medication should be started during the study, except the allowed rescue medication (paracetamol or tramadol, based on the experience from the DOLORES trial [53]). The baseline dose of pain medication may be reduced during the study, if required due to pain medication-related adverse events (AEs), and increased again up to baseline dose level if necessary. Further visits will be performed on Day 85 ± 4 days (V5) and Day 169 ± 4 days (V6). The primary analysis will be performed on data collected at V6. A follow-up visit will be performed on Day 183 ± 4 days (V7), approximately 2 weeks after the last intake of study medication (OPC 50 mg or PLC). Patients who discontinue early will be requested to attend an early discontinuation visit. At V6 (or early discontinuation visit, if applicable), the investigator will arrange the patient’s subsequent treatment (i.e. either prescribe further OPC or switch to another treatment).
Randomisation, blinding and allocation of treatment
At V1, each patient will be assigned in a chronological order via their electronic case report form to a unique patient number, which will be transferred by the site staff to an interactive web response system (IWRS). At V2b, after eligibility for entry into the treatment phase is confirmed, site staff will contact the IWRS to obtain the appropriate medication number. Randomisation will follow a 1:1 allocation rate (OPC 50 mg or PLC) and the randomisation list will be produced by the contract research organisation (Scope International AG, Mannheim, Germany) using the Statistical Analysis System (SAS) for Windows (SAS Institute Inc., Cary, NC, USA). The original list will be kept at the contract research organisation. Each patient’s investigational product will be determined by their randomisation number and corresponding medication number. OPC 50 mg and PLC capsules will be identical in size, colour, taste and appearance, and the packaging and labelling will not allow distinction between treatments. No person involved in conducting the study will have access to the randomisation code before the blind is officially broken. Unblinding will not occur unless there is an actual emergency and knowledge of the patient’s allocated treatment arm affects their treatment, in which case the individual treatment assignment for each patient will be available to the principal investigator/authorised delegate and responsible medical monitor via the IWRS. Patients with suspected unexpected serious adverse reactions (SUSARs) will be unblinded for regulatory reporting by the contract research organisation’s safety manager. Other study personnel and the investigators will receive blind information on the SUSAR until the study has been unblinded. The medication will be supplied by the sponsor (BIAL – Portela & Ca S.A., Coronado, Portugal) and the investigator/institution and/or a pharmacist or other appropriate individual (designated by the investigator/institution) will maintain records of delivery, inventory usage, and return any unused study medications. The investigator or an authorised delegate will be responsible for dispensing medication to the patients according to the dosage scheme and IWRS. At each study visit, site staff will dispense the appropriate amount of investigational product and rescue medication for each patient and for each treatment interval plus one extra week per 4 treatment weeks. At each visit, patients must bring back the study medication (including empty and partially empty containers) and accountability will be performed and documented.
Ethical considerations
The study will be conducted in accordance with: the Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Patients adopted by the General Assembly of the World Medical Association (2013); the applicable regulatory requirements of the participating countries; the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Harmonised Guideline – integrated addendum to ICH E6(R1) Guideline for Good Clinical Practice E6(R2); and with the European Commission Directives 2001/20/EC and 2005/28/EC, and EU Regulation No. 536/2014. The protocol will be submitted to national Independent Ethics Committee(s) and Competent Authorities and unconditional approval/favourable opinion must be obtained before the start of the study. All patients must provide written informed consent in order to participate in the study.
Data processing will be conducted by the contract research organisation. This will include, but is not limited to, producing the patient diary and electronic case report form, and setting up a relevant database and data transfer mechanisms, along with appropriate validation of data and resolution of queries. Clinical data will be collected in electronic form using an electronic data capture system. All clinical data will be recorded, processed, handled and stored without disclosing personal information of the patients so that the data can be accurately reported, interpreted and verified while the confidentiality of records and the personal data of the patients remain protected, in accordance with the applicable rules on personal data protection.
Study population
The study will be conducted in approximately 50 clinical sites in Germany, Italy, Portugal, Spain and the United Kingdom. Other countries and additional sites may be added, if required. Inclusion and exclusion criteria are outlined in Table 2.
Study assessments
An overview of study assessments is presented in Table 3 and the timing of these assessments is outlined in Fig. 2. Investigators will be trained on how to perform all assessments during each site initiation visit and at subsequent investigator meetings. Movement Disorder Society-sponsored Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) training certificates will be provided to all sites according to MDS procedures.

Timelines of study assessments. aV2 is divided in V2a and V2b. If ON/OFF diary entries are non-compliant at V2a, the patient will be re-trained on correct use of the diary and visit V2b will be postponed for 3–4 days. If diary completion is satisfactory at V2a, V2b is performed immediately on the same day. CGI-C, Clinical Global Impression of Change; DDCI, dopa decarboxylase inhibitor; EMD, early morning dystonia; KPPS, King’s Parkinson’s Disease Pain Scale; L-dopa, levodopa; MDS-NMS, Movement Disorder Society-sponsored Non-Motor rating Scale; MDS-UPDRS, Movement Disorder Society-sponsored Unified Parkinson’s Disease Rating Scale; PDQ-8, 8-item Parkinson’s Disease Questionnaire; PGI-C, Patient’s Global Impression of Change; PSV, post-study visit; V, visit
Efficacy
The primary efficacy endpoint is change from baseline in Domain 3 (fluctuation-related pain) of the KPPS. The KPPS evaluates the burden and characterises various phenotypes of pain in PD. It comprises seven domains including a total of 14 items. Each item is scored by severity (0–3) multiplied by frequency (0–4), resulting in subscores of 0–12. The total KPPS score (0–168) represents the symptomatic burden by pain [35].
The key secondary efficacy endpoint is change from baseline in Domain B (anxiety) of the Movement Disorder Society-sponsored Non-Motor rating Scale (MDS-NMS). The MDS-NMS comprises 13 domains covering a range of key PD- and treatment-related NMS, and a subscale for NMF that assesses changes in NMS in relation to timing of anti-PD medications across eight domains [54, 55]. Additional secondary efficacy endpoints comprise of other domains and total scores of the KPPS and MDS-NMS, change from baseline in the MDS-UPDRS Parts III and IV, change from baseline in the Parkinson’s Disease Questionnaire (PDQ-8), Clinical Global Impression of Change (CGI-C), Patient’s Global Impression of Change (PGI-C), change from baseline in functional status via Hauser’s PD diary, changes from baseline in morning dystonia, and use of rescue medication (Table 3).
The MDS-UPDRS is a revision of the UPDRS originally developed in the 1980s, and evaluates various aspects of PD; it consists of four parts [56]: Parts IA and IB, non-motor aspects of experiences of daily living; Part II, motor aspects of experiences of daily living; Part III, motor examination; and Part IV, motor complications. The PDQ-8 (a short form of the PDQ-39) is a patient-reported outcome that assesses eight aspects of functioning and well-being that are usually adversely affected by PD: mobility, activities of daily living, emotional well-being, stigma, social support, cognition, communication, and bodily discomfort. It rates overall health status by providing a single score ranging from 0 (good health) to 100 (poor health) [57]. The CGI-C and PGI-C are, respectively, investigator and patient assessments of how much a patient’s overall status has improved or worsened since the start of the study, comprising a 7-point scale: (1, ‘very much improved’; 2, ‘much improved’; 3, ‘minimally improved’; 4, ‘no change’; 5, ‘minimally worse’; 6, ‘much worse’; 7, ‘very much worse’). The Hauser’s PD diary is a patient record of their mobility during each 30-min period, categorised as: asleep; OFF time; ON time without dyskinesia; ON time with non-troublesome dyskinesia; or ON time with troublesome dyskinesia. When assessing changes from baseline in morning dystonia, the investigator will ask the patient if they experienced any morning dystonia within the last week (based on item 35 of the former UPDRS version). The amount and frequency of intake of rescue medication (paracetamol or tramadol) will be recorded by patients in a diary.
Safety assessments
Safety assessments include the incidence of treatment-emergent AEs (TEAEs), and changes from baseline in vital signs, physical and neurological examinations and routine laboratory parameters (Table 3; Fig. 2). At each study visit, the investigator will ask the patient in a non-leading manner about the state of their health in order to illicit information on TEAEs that may have occurred since the last visit. Any clinically significant observations made during the visit also constitute TEAEs. TEAEs will be documented as soon as possible in the electronic patient report form. The following information will also be specified: date/time of onset of TEAE; action taken with the investigational product; other actions taken; outcome of TEAE; seriousness of TEAE; severity of TEAE (mild, moderate, severe); and causal relationship of TEAE to investigational product (unrelated, unlikely, possible, probably, definite).
Sample size calculation
For the primary efficacy endpoint (change from baseline in Domain 3 of KPPS), a difference to PLC of 3.0 is regarded as clinically meaningful. From a former study [53], a standard deviation (SD) of 5.8 can be assumed. With a two-sided significance α of 0.05, a power of 80%, a 1:1 treatment allocation ratio and with the above-mentioned assumptions, 2 × 60 = 120 evaluable patients are required. Assuming a drop-out rate of 15%, a total of 140 patients need to be randomised. Randomisation will follow a 1:1 allocation rate (OPC 50 mg or PLC).
Statistical methodology
Efficacy assessments will be analysed for the Full Analysis Set, defined as all patients who are randomised and who have at least one measurement of the primary efficacy assessment. For sensitivity purposes, efficacy assessments will additionally be analysed for the Per-Protocol Set, defined as all patients included in the Full Analysis Set who have no major protocol deviations that could influence the primary efficacy assessment. The primary efficacy endpoint will be analysed using analysis of covariance (ANCOVA), with treatment as a fixed factor and baseline KPPS as a covariate, to demonstrate superiority of OPC 50 mg against PLC. Secondary efficacy endpoints will be analysed in an exploratory manner by treatment arm using appropriate parametric and non-parametric statistical methods. Descriptive statistics, including 95% confidence intervals, will be presented per treatment arm.
Safety assessments will be analysed for the Safety Set, defined as all patients who take at least one dose of investigational product. TEAEs will be summarised in terms of the number and percentages of patients with TEAEs. Vital signs and laboratory parameters will be summarised using summary statistics of absolute values and changes from baseline. Summary statistics and shift tables will be presented for physical and neurological examinations. Demographic and baseline characteristics will be presented using descriptive statistics. The statistical analysis plan will be carried out by biostatisticians from the contract research organisation.
Current status
The first patient was enrolled in February 2021 in the UK. The recruitment window is now open and the last patient is expected to complete the study by late 2022. Timelines might be impacted by recurring COVID-19-related lockdowns impairing the access of patients to healthcare facilities.
Discussion
Pain has a major impact on the quality of life of patients with PD [23, 58,59,60] and nociceptive pain accounts for the majority of reported pain in PD [22]. Since pain modulation involves striatal dopamine D2 receptors [61], pain associated with end-of-dose motor fluctuations may be alleviated through optimisation of dopaminergic therapy [12, 21, 22, 29].
Management of pain, among many other non-motor aspects of PD, remains a key unmet need [62] and there is currently a lack of robust data on the management of pain in PD patients with end-of-dose motor fluctuations. Previous studies in this setting have notable limitations, as well as varying both in the tools used to measure pain and the types of pain assessed. The Phase II PANDA trial was the first randomised controlled trial to specifically assess treatment for PD-associated pain [34]. Eligible patients were randomised to receive either prolonged-release oxycodone-naloxone or placebo. The types of pain patients experienced at baseline included musculoskeletal pain (73% in active arm), nocturnal pain (35%), fluctuation-related pain (32%) and PD-related chronic pain (26%). There was no significant difference between treatment arms in the average 24-h pain score at 16 weeks (primary endpoint). However, the measure used to assess pain was a general pain scale (a Likert scale) and levodopa was used more frequently as a rescue treatment in the placebo arm, both of which factors might have affected the results [34]. The double-blind, exploratory DOLORES trial was the first to investigate the effect of a dopamine agonist (rotigotine; administered as a transdermal patch) on PD-associated pain as primary outcome [53]. The types of pain patients experienced at baseline included musculoskeletal pain (51% in active arm), neuropathic pain (23%) and dystonic pain (14%). Although the findings suggested that rotigotine may improve PD-associated chronic pain in patients with advanced-stage PD, the trial was not powered to detect statistically significant treatment differences, due to the small sample size [53]. Safinamide (an agent with multiple modes of action, including monoamine oxidase-B inhibition) was shown to significantly reduce the need for pain medication, and to significantly improve two out of three PDQ-39 pain-related items, in comparison with placebo, when added to existing levodopa-based therapy [13]. However, these results were based on a post-hoc analysis of two previous trials and must therefore be interpreted with caution.
The design of the OCEAN study will address the current lack of reliable evidence for levodopa-based therapy in the treatment of PD-associated pain. OCEAN features recent validated PD pain- and non-motor-specific scales (such as the KPPS and the MDS-NMS), which might help to record dimensions of PD-associated pain that have not been previously studied. For instance, this study may allow the detection of potential associations between pain and other NMS, such as depression, anxiety and insomnia, and dysautonomic symptoms. The concomitant use of ON/OFF diaries with these scales may also allow a deeper understanding of pain and other NMS, such as the key secondary endpoint anxiety (as assessed by change from baseline in Domain B of the MDS-NMS), during both the OFF and ON states. Anxiety problems, including OFF-period anxiety, are highly prevalent in PD and greatly impact quality of life [63, 64]. Although some data suggest that anxiety symptoms inversely correlate with motor improvement induced by oral levodopa [65], this has been a neglected area in clinical investigation [30]. Placebo is known to activate dopamine receptors and to induce dopamine-like effects in PD [66,67,68,69], which are often still apparent in studies at 3 months [34, 47, 48], tending to wane by the 6-month mark [70, 71]. The 6-month course of OCEAN and its double-blind design might therefore help to disentangle the placebo dopamine-mediated effect from the true pharmacological benefit of OPC, especially when evaluating pain.
In summary, the OCEAN study will provide valuable information on whether the use of adjunctive OPC 50 mg treatment can improve fluctuation-associated pain in PD patients with end-of-dose motor fluctuations. The data will address the current lack of Level 1 evidence for the recommendation of strategies to manage aspects of pain in PD.
Availability of data and materials
Protocol details are available at www.clinicaltrialsregister.eu (EudraCT number 2020–001175-32). In line with EFPIA and PhRMA guiding principles, BIAL undertakes to share, upon request, anonymised patient-level, study-level clinical trial data (analysable data sets), and other information (such as protocols) from clinical trials in patients for medicines and indications approved in the United States (US) and the European Union (EU), to qualified researchers as necessary for conducting legitimate research.
Abbreviations
- AE:
-
Adverse event
- ANCOVA:
-
Analysis of covariance
- CGI-C:
-
Clinical Global Impression of Change
- COMT:
-
Catechol-O-methyltransferase
- COMTi:
-
Catechol-O-methyltransferase inhibitor
- DDC:
-
Dopa decarboxylase
- DDCi:
-
Dopa decarboxylase inhibitor
- EC:
-
European Commission
- ICH:
-
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- IWRS:
-
Interactive web response system
- KPPS:
-
King’s Parkinson’s Disease Pain Scale
- L-dopa:
-
Levodopa
- MDS-NMS:
-
Movement Disorder Society-sponsored Non-Motor rating Scale
- MDS-UPDRS:
-
Movement Disorder Society-sponsored Unified Parkinson’s Disease Rating Scale
- NMF:
-
Non-motor fluctuations
- NMS:
-
Non-motor symptoms
- NMSS:
-
Non-Motor Symptoms Scale
- OCEAN:
-
OpiCapone Effect on motor fluctuations and pAiN study
- OPC:
-
Opicapone
- PD:
-
Parkinson’s disease
- PDQ-8:
-
8-item Parkinson’s Disease Questionnaire
- PDQ-39:
-
39-item Parkinson’s Disease Questionnaire
- PGI-C:
-
Patient’s Global Impression of Change
- PLC:
-
Placebo
- SAS:
-
Statistical Analysis System
- SD:
-
Standard deviation
- SUSAR:
-
Suspected unexpected serious adverse reaction
- TEAE:
-
Treatment-emergent adverse event
- UPDRS:
-
Unified Parkinson’s Disease Rating Scale
- USA:
-
United States of America
- V:
-
Visit
References
Parkinson J. An essay on the shaking palsy. London: Sherwood, Neely, and Jones; 1817.
Stern G. Did parkinsonism occur before 1817? J Neurol Neurosurg Psychiatry. 1989;Suppl(Suppl):11–2. https://doi.org/10.1136/jnnp.52.suppl.11.
Bereczki D. The description of all four cardinal signs of Parkinson's disease in a Hungarian medical text published in 1690. Parkinsonism Relat Disord. 2010;16:290–3. https://doi.org/10.1016/j.parkreldis.2009.11.006.
Ray Chaudhuri K, Poewe W, Brooks D. Motor and nonmotor complications of levodopa: phenomenology, risk factors, and imaging features. Mov Disord. 2018;33:909–19. https://doi.org/10.1002/mds.27386.
Storch A, Schneider CB, Wolz M, Stürwald Y, Nebe A, Odin P, et al. Nonmotor fluctuations in Parkinson disease: severity and correlation with motor complications. Neurology. 2013;80:800–9. https://doi.org/10.1212/WNL.0b013e318285c0ed.
Kleiner-Fisman G, Martine R, Lang AE, Stern MB. Development of a non-motor fluctuation assessment instrument for Parkinson disease. Parkinsons Dis. 2011;2011:292719. https://doi.org/10.4061/2011/292719.
Brun L, Lefaucheur R, Fetter D, Derrey S, Borden A, Wallon D, et al. Non-motor fluctuations in Parkinson's disease: prevalence, characteristics and management in a large cohort of parkinsonian outpatients. Clin Neurol Neurosurg. 2014;127:93–6. https://doi.org/10.1016/j.clineuro.2014.10.006.
Martínez-Fernández R, Schmitt E, Martinez-Martin P, Krack P. The hidden sister of motor fluctuations in Parkinson's disease: a review on nonmotor fluctuations. Mov Disord. 2016;31:1080–94. https://doi.org/10.1002/mds.26731.
Grosset D, Taurah L, Burn DJ, MacMahon D, Forbes A, Turner K, et al. A multicentre longitudinal observational study of changes in self reported health status in people with Parkinson's disease left untreated at diagnosis. J Neurol Neurosurg Psychiatry. 2007;78:465–9. https://doi.org/10.1136/jnnp.2006.098327.
Antonini A, Barone P, Marconi R, Morgante L, Zappulla S, Pontieri FE, et al. The progression of non-motor symptoms in Parkinson's disease and their contribution to motor disability and quality of life. J Neurol. 2012;259:2621–31. https://doi.org/10.1007/s00415-012-6557-8.
Nègre-Pagès L, Regragui W, Bouhassira D, Grandjean H, Rascol O. DoPaMiP study group. Chronic pain in Parkinson's disease: the cross-sectional French DoPaMiP survey. Mov Disord. 2008;23:1361–9. https://doi.org/10.1002/mds.22142.
Antonini A, Tinazzi M, Abbruzzese G, Berardelli A, Chaudhuri KR, Defazio G, et al. Pain in Parkinson's disease: facts and uncertainties. Eur J Neurol. 2018;25:917–e69. https://doi.org/10.1111/ene.13624.
Cattaneo C, Kulisevsky J, Tubazio V, Castellani P. Long-term efficacy of safinamide on Parkinson's disease chronic pain. Adv Ther. 2018;35:515–22. https://doi.org/10.1007/s12325-018-0687-z.
Buhmann C, Kassubek J, Jost WH. Management of pain in Parkinson's disease. J Parkinsons Dis. 2020;10(Suppl 1):S37–48. https://doi.org/10.3233/JPD-202069.
Marques A, Brefel-Courbon C. Chronic pain in Parkinson's disease: clinical and pathophysiological aspects. Rev Neurol (Paris). 2021;177:394–9. https://doi.org/10.1016/j.neurol.2020.06.015.
Ghosh P, Imbriani P, Caputi N, Natoli S, Schirinzi T, Di Lazzaro G, et al. A dual Centre study of pain in Parkinson's disease and its relationship with other non-motor symptoms. J Parkinsons Dis. 2020;10:1817–25. https://doi.org/10.3233/JPD-202088.
Polli A, Weis L, Biundo R, Thacker M, Turolla A, Koutsikos K, et al. Anatomical and functional correlates of persistent pain in Parkinson's disease. Mov Disord. 2016;31:1854–64. https://doi.org/10.1002/mds.26826.
Ford B. Pain in Parkinson's disease. Mov Disord. 2010;25(Suppl 1):S98–103. https://doi.org/10.1002/mds.22716.
Valkovic P, Minar M, Singliarova H, Harsany J, Hanakova M, Martinkova J, et al. Pain in Parkinson's disease: a cross-sectional study of its prevalence, types, and relationship to depression and quality of life. PLoS One. 2015;10:e0136541. https://doi.org/10.1371/journal.pone.0136541.
Edinoff A, Sathivadivel N, McBride T, Parker A, Okeagu C, Kaye AD, et al. Chronic pain treatment strategies in Parkinson's disease. Neurol Int. 2020;12:61–76. https://doi.org/10.3390/neurolint12030014.
Brefel-Courbon C, Ory-Magne F, Thalamas C, Payoux P, Rascol O. Nociceptive brain activation in patients with neuropathic pain related to Parkinson's disease. Parkinsonism Relat Disord. 2013;19:548–52. https://doi.org/10.1016/j.parkreldis.2013.02.003.
Rukavina K, Leta V, Sportelli C, Buhidma Y, Duty S, Malcangio M, et al. Pain in Parkinson's disease: new concepts in pathogenesis and treatment. Curr Opin Neurol. 2019;32:579–88. https://doi.org/10.1097/WCO.0000000000000711.
Truini A, Frontoni M, Cruccu G. Parkinson's disease related pain: a review of recent findings. J Neurol. 2013;260:330–4. https://doi.org/10.1007/s00415-012-6754-5.
Rukavina K, Cummins TM, Ray CK, Kirtsy B. Pain in Parkinson's disease: mechanism-based treatment strategies. Curr Opin Support Palliat Care. 2021;15:108–15. https://doi.org/10.1097/SPC.0000000000000546.
Karnik V, Farcy N, Zamorano C, Bruno V. Current status of pain Management in Parkinson's disease. Can J Neurol Sci. 2020;47:336–43. https://doi.org/10.1017/cjn.2020.13.
Rodríguez-Violante M, Alvarado-Bolaños A, Cervantes-Arriaga A, Martinez-Martin P, Rizos A, Chaudhuri KR. Clinical determinants of Parkinson's disease-associated pain using the King's Parkinson's disease pain scale. Mov Disord Clin Pract. 2017;4:545–51. https://doi.org/10.1002/mdc3.12469.
Navratilova E, Xie JY, Meske D, Qu C, Morimura K, Okun A, et al. Endogenous opioid activity in the anterior cingulate cortex is required for relief of pain. J Neurosci. 2015;35:7264–71. https://doi.org/10.1523/JNEUROSCI.3862-14.2015.
García-Ramírez DL, Calvo JR, Hochman S, Quevedo JN. Serotonin, dopamine and noradrenaline adjust actions of myelinated afferents via modulation of presynaptic inhibition in the mouse spinal cord. PLoS One. 2014;9:e89999. https://doi.org/10.1371/journal.pone.0089999.
Brefel-Courbon C, Payoux P, Thalamas C, Ory F, Quelven I, Chollet F, et al. Effect of levodopa on pain threshold in Parkinson's disease: a clinical and positron emission tomography study. Mov Disord. 2005;20:1557–63. https://doi.org/10.1002/mds.20629.
Seppi K, Chaudhuri KR, Coelho M, Fox SH, Katzenschlager R, Perez Lloret S, et al. The collaborators of the Parkinson's disease update on non-motor symptoms study group on behalf of the movement disorders society evidence-based medicine committee. Update on treatments for nonmotor symptoms of Parkinson's disease-an evidence-based medicine review. Mov Disord. 2019;34:180–98. https://doi.org/10.1002/mds.27602.
Bannister K, Dickenson AH. What do monoamines do in pain modulation? Curr Opin Support Palliat Care. 2016;10:143–8. https://doi.org/10.1097/SPC.0000000000000207.
Jung YJ, Kim HJ, Jeon BS, Park H, Lee WW, Paek SH. An 8-year follow-up on the effect of subthalamic nucleus deep brain stimulation on pain in Parkinson disease. JAMA Neurol. 2015;72:504–10. https://doi.org/10.1001/jamaneurol.2015.8.
Dafsari HS, Martinez-Martin P, Rizos A, Trost M, Dos Santos Ghilardi MG, Reddy P, et al. EUROPAR and the international Parkinson and movement disorders society non-motor Parkinson's disease study group. EuroInf 2: subthalamic stimulation, apomorphine, and levodopa infusion in Parkinson's disease. Mov Disord. 2019;34:353–65. https://doi.org/10.1002/mds.27626.
Trenkwalder C, Chaudhuri KR, Martinez-Martin P, Rascol O, Ehret R, Vališ M, et al. PANDA study group. Prolonged-release oxycodone-naloxone for treatment of severe pain in patients with Parkinson's disease (PANDA): a double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2015;14:1161–70. https://doi.org/10.1016/S1474-4422(15)00243-4.
Chaudhuri KR, Rizos A, Trenkwalder C, Rascol O, Pal S, Martino D, et al. EUROPAR and the IPMDS non motor PD study group. King's Parkinson's disease pain scale, the first scale for pain in PD: an international validation. Mov Disord. 2015;30:1623–31. https://doi.org/10.1002/mds.26270.
Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson's disease: an old drug still going strong. Clin Interv Aging. 2010;5:229–38. https://doi.org/10.2147/cia.s6456.
Kiss LE, Ferreira HS, Torrão L, Bonifácio MJ, Palma PN, Soares-da-Silva P, et al. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53:3396–411. https://doi.org/10.1021/jm1001524.
Almeida L, Rocha JF, Falcão A, Palma PN, Loureiro AI, Pinto R, et al. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013;52:139–51. https://doi.org/10.1007/s40262-012-0024-7.
Ouma S, Fukae J, Fujioka S, Yamamoto S, Hatano T, Yoritaka A, et al. The risk factors for the wearing-off phenomenon in Parkinson's disease in Japan: a cross-sectional, multicenter study. Intern Med. 2017;56:1961–6. https://doi.org/10.2169/internalmedicine.56.7667.
Cheon SM, Park MJ, Kim WJ, Kim JW. Non-motor off symptoms in Parkinson's disease. J Korean Med Sci. 2009;24:311–4. https://doi.org/10.3346/jkms.2009.24.2.311.
Sung S, Farrell M, Vijiaratnam N, Evans AH. Pain and dyskinesia in Parkinson's disease may share common pathophysiological mechanisms - an fMRI study. J Neurol Sci. 2020;416:116905. https://doi.org/10.1016/j.jns.2020.116905.
Sung S, Vijiaratnam N, Chan DWC, Farrell M, Evans AH. Parkinson disease: a systemic review of pain sensitivities and its association with clinical pain and response to dopaminergic stimulation. J Neurol Sci. 2018;395:172–206. https://doi.org/10.1016/j.jns.2018.10.013.
Müller T. Catechol-O-methyltransferase inhibitors in Parkinson's disease. Drugs. 2015;75:157–74. https://doi.org/10.1007/s40265-014-0343-0.
Montioli R, Voltattorni CB, Bertoldi M. Parkinson's disease: recent updates in the identification of human dopa decarboxylase inhibitors. Curr Drug Metab. 2016;17:513–8. https://doi.org/10.2174/138920021705160324170558.
Scott LJ. Opicapone: a review in Parkinson's disease. Drugs. 2016;76:1293–300. https://doi.org/10.1007/s40265-016-0623-y.
Fabbri M, Ferreira JJ, Lees A, Stocchi F, Poewe W, Tolosa E, et al. Opicapone for the treatment of Parkinson's disease: a review of a new licensed medicine. Mov Disord. 2018;33:1528–39. https://doi.org/10.1002/mds.27475.
Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P. Bi-Park 1 investigators. Opicapone as an adjunct to levodopa in patients with Parkinson's disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2016;15:154–65. https://doi.org/10.1016/S1474-4422(15)00336-1.
Lees AJ, Ferreira J, Rascol O, Poewe W, Rocha JF, McCrory M, et al. BIPARK-2 study investigators. Opicapone as adjunct to levodopa therapy in patients with Parkinson disease and motor fluctuations: a randomized clinical trial. JAMA Neurol. 2017;74:197–206. https://doi.org/10.1001/jamaneurol.2016.4703.
Ongentys® Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product-information/ongentys-epar-product-information_en.pdf. Accessed 25 Nov 2021.
Ongentys® Highlights of Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212489s000lbl.pdf. Accessed 25 Nov 2021.
Oliveira C, Lees A, Ferreira J, Lopes N, Costa R, Pinto R, et al. Evaluation of non-motor symptoms in opicapone treated Parkinson’s disease patients: results from a double-blind, randomized, placebo-controlled study and open-label extension. Eur J Neurol. 2015;22(Suppl 1):191 abstract P1236.
Reichmann H, Lees A, Rocha JF, Magalhães D, Soares-da-Silva P, OPTIPARK investigators. Effectiveness and safety of opicapone in Parkinson's disease patients with motor fluctuations: the OPTIPARK open-label study. Transl Neurodegener. 2020;9:9. https://doi.org/10.1186/s40035-020-00187-1.
Rascol O, Zesiewicz T, Chaudhuri KR, Asgharnejad M, Surmann E, Dohin E, et al. A randomized controlled exploratory pilot study to evaluate the effect of rotigotine transdermal patch on Parkinson's disease-associated chronic pain. J Clin Pharmacol. 2016;56:852–61. https://doi.org/10.1002/jcph.678.
Martinez-Martin P, Schrag A, Weintraub D, Rizos A, Rodriguez-Blazquez C, Chaudhuri KR. IPMDS non motor PD study group. Pilot study of the International Parkinson and Movement Disorder Society-sponsored non-motor rating scale (MDS-NMS). Mov Disord Clin Pract. 2019;6:227–34. https://doi.org/10.1002/mdc3.12728.
Chaudhuri KR, Schrag A, Weintraub D, Rizos A, Rodriguez-Blazquez C, Mamikonyan E, et al. The movement disorder society nonmotor rating scale: initial validation study. Mov Disord. 2020;35:116–33. https://doi.org/10.1002/mds.27862.
Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement Disorder Society UPDRS revision task force. Movement Disorder Society-sponsored revision of the unified Parkinson's disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–70. https://doi.org/10.1002/mds.22340.
Jenkinson C, Fitzpatrick R, Peto V, Greenhall R, Hyman N. The PDQ-8: development and validation of a short-form Parkinson's disease questionnaire. Psychol Health. 1997;12:805–14. https://doi.org/10.1080/08870449708406741.
Chaudhuri KR, Healy DG, Schapira AH. National Institute for clinical excellence. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 2006;5:235–45. https://doi.org/10.1016/S1474-4422(06)70373-8.
Ozturk EA, Gundogdu I, Kocer B, Comoglu S, Cakci A. Chronic pain in Parkinson's disease: frequency, characteristics, independent factors, and relationship with health-related quality of life. J Back Musculoskelet Rehabil. 2016. https://doi.org/10.3233/BMR-160720.
Choi SM, Kim BC, Jung HJ, Yoon GJ, Kang KW, Choi KH, et al. Impact of pain and pain subtypes on the quality of life of patients with Parkinson's disease. J Clin Neurosci. 2017;45:105–9. https://doi.org/10.1016/j.jocn.2017.08.002.
Hagelberg N, Jääskeläinen SK, Martikainen IK, Mansikka H, Forssell H, Scheinin H, et al. Striatal dopamine D2 receptors in modulation of pain in humans: a review. Eur J Pharmacol. 2004;500:187–92. https://doi.org/10.1016/j.ejphar.2004.07.024.
LeWitt PA, Chaudhuri KR. Unmet needs in Parkinson disease: motor and non-motor. Parkinsonism Relat Disord. 2020;80(Suppl 1):S7–S12. https://doi.org/10.1016/j.parkreldis.2020.09.024.
Pontone GM, Dissanayka N, Apostolova L, Brown RG, Dobkin R, Dujardin K, et al. Report from a multidisciplinary meeting on anxiety as a non-motor manifestation of Parkinson's disease. NPJ Parkinsons Dis. 2019;5:30. https://doi.org/10.1038/s41531-019-0102-8.
Aquino CC, Fox SH. Clinical spectrum of levodopa-induced complications. Mov Disord. 2015;30:80–9. https://doi.org/10.1002/mds.26125.
Kulisevsky J, Pascual-Sedano B, Barbanoj M, Gironell A, Pagonabarraga J, García-Sánchez C. Acute effects of immediate and controlled-release levodopa on mood in Parkinson's disease: a double-blind study. Mov Disord. 2007;22:62–7. https://doi.org/10.1002/mds.21205.
de la Fuente-Fernández R, Schulzer M, Stoessl AJ. The placebo effect in neurological disorders. Lancet Neurol. 2002;1:85–91. https://doi.org/10.1016/s1474-4422(02)00038-8.
Benedetti F. Placebo effects: from the neurobiological paradigm to translational implications. Neuron. 2014;84:623–37. https://doi.org/10.1016/j.neuron.2014.10.023.
Colloca L. The placebo effect in pain therapies. Annu Rev Pharmacol Toxicol. 2019;59:191–211. https://doi.org/10.1146/annurev-pharmtox-010818-021542.
Lou JS. Placebo responses in Parkinson's disease. Int Rev Neurobiol. 2020;153:187–211. https://doi.org/10.1016/bs.irn.2020.03.031.
Borgohain R, Szasz J, Stanzione P, Meshram C, Bhatt M, Chirilineau D, et al. Study 016 investigators. Randomized trial of safinamide add-on to levodopa in Parkinson's disease with motor fluctuations. Mov Disord. 2014;29:229–37. https://doi.org/10.1002/mds.25751.
Hattori N, Tsuboi Y, Yamamoto A, Sasagawa Y, Nomoto M. ME2125-3 study group. Efficacy and safety of safinamide as an add-on therapy to L-DOPA for patients with Parkinson's disease: a randomized, double-blind, placebo-controlled, phase II/III study. Parkinsonism Relat Disord. 2020;75:17–23. https://doi.org/10.1016/j.parkreldis.2020.04.012.
Acknowledgements
Editorial assistance was provided by John Scopes of mXm Medical Communications and funded by Bial – Portela & Cª, S.A.
Funding
The study design, data collection/analysis/interpretation and editorial assistance were funded by Bial – Portela & Cª, S.A. Employees of Bial – Portela & Ca, S.A. participated in the design of the study and in the collection, analysis and interpretation of data.
Author information
Authors and Affiliations
Contributions
All authors participated in the study design. JFR, DM, RC and PSS are participating in the study implementation. All authors were involved in the initial ideas presented in the introduction and discussion of this article. KRC wrote a major part the first draft of the manuscript; PO, JJF, AA, OR, MMK, AS and KB made substantial contributions to the writing and revising of the manuscript; and JFR, DM, RC and PSS provided critical review. All authors approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol was submitted to national Independent Ethics Committees and Competent Authorities and unconditional approval/favourable opinion must be obtained before the start of the study. All patients must provide written informed consent in order to participate in the study. The study was submitted to and approved by the following Independent Ethics Committees: in Spain, CEIm Hospital Clínic de Barcelona to cover all sites; in Portugal, Comissão de Ética para a Investigação Clínica to cover all sites; in the UK, London-Hampstead Research Ethics Committee and the Health Research Authority to cover all sites; in Germany, Ethikkommission bei der Medizinischen Fakultät der LMU München, Ethik-Kommission des Landes Berlin, Ethik-Kommission der Bayerischen Landesärztekammer, Ethikkommission der Fakultät für Medizin der Technischen Universität München, Ethikkommission der Landesärztekammer Thüringen, Universität Duisburg-Essen Ethik-Kommission and Ethik-Kommission bei der Landesärztekammer Hessen; in Italy, Comitato Etico per la Sperimentazione Clinica della Provincia di Padova, Comitato Etico per la sperimentazione clinica della provincia di Venezia e IRCCS San Camillo, Comitato Etico referente Area Pavia- Direzione Scientifica Comitato Etico ASL Brindisi, Comitato Etico Interaziendale Novara, Comitato Etico Indipendente locale - AOU Consorziale Policlinico di Bari and Comitato Etico di Brescia Comitato Etico per la Ricerca Biomedica delle Province di Chieti e Pescara e dell’Università degli Studi “G. D’Annunzio” di Chieti-Pescara.
Consent for publication
Not applicable.
Competing interests
KRC has participated in advisory boards for AbbVie, UCB, GKC, Bial, Cynapsus, Lobsor, STADA, Medtronic, Zambon, Profile, Sunovion, Roche, Theravance, Scion, Britannia, Acadia, and 4D; has received honoraria for lectures from AbbVie, Britannia, UCB, Zambon, Novartis, Boehringer Ingelheim, Bial, Kyowa Kirin, and SK Pharma; has received investigator-initiated grants from Britannia, AbbVie, UCB, GKC, and Bial; and has received academic grants from the EU, IMI EU, Horizon 2020, Parkinson’s UK, National Institute for Health Research, Parkinson’s Disease Non Motor Group, Kirby Laing Foundation, NPF, Medical Research Council, and Wellcome Trust.
PO has received honoraria for advice and lectures for AbbVie, Bial, Britannia, Lobsor, Nordic Infucare and Zambon, and has received grants from AbbVie.
JJF has provided consultancy for Ipsen, GlaxoSmithKline, Novartis, Teva, Lundbeck, Solvay, Abbott, BIAL, Merck-Serono and Merz; and has received grants from GlaxoSmithKline, Grunenthal, Teva and Fundação MSD.
AA has received compensation for consultancy and speaker-related activities from UCB, Boehringer Ingelheim, Britannia, AbbVie, Zambon, Bial, NeuroDerm, Theravance Biopharma, Roche; he receives research support from Chiesi Pharmaceuticals, Lundbeck, Horizon 2020 - Grant 825785, Horizon 2020 - Grant 101016902, Ministry of Education University and Research (MIUR) Grant ARS01_01081, Cariparo Foundation. He serves as consultant for Boehringer Ingelheim for legal cases on pathological gambling; owns Patent WO2015110261-A1; and owns shares in PD Neurotechnology Limited.
OR has participated in advisory boards and/or provided consultancy for AbbVie, Adamas, Acorda, Addex, AlzProtect, ApoPharma, AstraZeneca, Axovant, Bial, Biogen, Britannia, Buckwang, CereSpir, Clevexel, Denali, INC Research, IPMDS, Lundbeck, Lupin, Merck, MundiPharma, NeurATRIS, NeuroDerm, Novartis, ONO Pharma, Osmotica, Parexel, Pfizer, Prexton Therapeutics, Quintiles, Roche, Sanofi, Servier, Sunovion, Theranexus, Takeda, Teva, UCB, Vectura, Watermark Research, XenoPort, XO, Zambon; received grants from Agence Nationale de la Recherche (ANR), CHU de Toulouse, France-Parkinson, INSERM-DHOS Recherche Clinique Translationnelle, MJFox Foundation, Programme Hospitalier de Recherche Clinique, European Commission (FP7, H2020), Cure Parkinson’s UK; and received a grant to participate in a symposium and contribute to the review of an article by the International Parkinson and Movement Disorder Society.
MMK has received honoraria from AbbVie, Zambon, Bial and the International Parkinson and Movement Disorder Society.
AS has received funding from the Deutsche Forschungsgemeinschaft (German Research Association) and the Helmholtz-Association, outside the submitted presented work. He has received honoraria for presentations/advisory boards/consultations from Desitin, Global Kinetics, Lobsor, STADA, Bial, RG Gesellschaft, and AbbVie, outside the submitted presented work. He has received royalties from Kohlhammer Verlag and Elsevier Press. He serves as an editorial board member of Stem Cells and Stem Cells International.
KB has no competing interests.
PSS is an employee of Bial – Portela & Cª, S.A.
RC is an employee of Bial – Portela & Cª, S.A.
DM is an employee of Bial – Portela & Cª, S.A.
JFR is an employee of Bial – Portela & Cª, S.A.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chaudhuri, K.R., Odin, P., Ferreira, J.J. et al. Opicapone versus placebo in the treatment of Parkinson’s disease patients with end-of-dose motor fluctuation-associated pain: rationale and design of the randomised, double-blind OCEAN (OpiCapone Effect on motor fluctuations and pAiN) trial. BMC Neurol 22, 88 (2022). https://doi.org/10.1186/s12883-022-02602-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-022-02602-8