
Abstract
Background
As investigations of disease modifying drugs aim to slow down progression of Alzheimer’ disease (AD) biomarkers to reliably track disease progression gain more importance. This is especially important as clinical symptoms, including psychometric measures, are only modestly associated with the underlying disease pathology, in particular at the pre-dementia stages. The decision which biomarkers to choose in clinical trials is crucial and depends on effect size. However, longitudinal studies of multiple biomarkers in parallel that allow direct comparison on effect size are scarce.
Methods
We calculated effect size and minimal sample size for three common imaging biomarkers of AD, namely amyloid deposition measured with PiB-PET, neuronal dysfunction measured with FDG-PET and cortical thickness measured with MRI in a prospective 24-month follow-up study in a monocentric cohort of early AD.
Results
Post hoc power calculation revealed large effect sizes of Cohen’s d for PiB-PET and cortical thickness and a small effect size for FDG-PET (1.315, 0.914, and 0.341, respectively). Accordingly, sample sizes for PiB-PET and cortical thickness required significantly smaller sample sizes than FDG-PET to reliably detect statistically significant changes after 24 months in early AD (n = 7, n = 12, and n = 70, respectively).
Conclusion
Amyloid imaging with PET and measuring cortical thickness with MRI are suitable biomarkers to detect disease progression in early AD within a small sample.
Similar content being viewed by others

Background
While there is still no definite treatment for the cause of Alzheimer’s disease (AD), modifying therapies that aim to slow down disease progression are under investigation. Primary endpoints in these trials are usually measures of cognitive functions such as the Alzheimer’s Disease Assessment Scale - Cognitive Subscale (ADAS-cog) [1] or the Clinical Dementia Rating Scale (CDR) [2,3,4,5]. However, the progression of clinical symptoms varies among patients with AD and is not always closely associated with the progression of underlying disease, namely beta-amyloid (Aβ) and tau deposition in conjugation with synaptic and neuronal loss [6]. The presentation of clinical symptoms could be modified by brain cognitive reserve capacity [7] and other factors, for example ceiling and floor effects of psychometric tests such as the ADAS-Cog [8]. Thus, to assess the efficacy of disease modifying therapies, biomarkers reflecting the characteristic histopathological features of AD might be more suitable than psychometric tests [9]. Biomarkers for Aβ accumulation are positive uptake of an amyloid tracer, such as 11C-Pittsburgh compound B (PiB), on positron emission tomography (PET) and decreased Aβ1–42 in cerebrospinal fluid (CSF) [6]. Biomarkers for tau pathology include tau PET and CSF p-tau. Biomarkers for neuronal dysfunction include specific regions with reduced 18F-fluordeoxyglucose (FDG) uptake on PET, and for neuronal loss atrophy on magnetic resonance imaging (MRI) and elevated t-tau protein in CSF [6]. The vast majority of these biomarkers have been studied in terms of specificity, validity, change over time, and correlation with clinical symptoms and other biomarkers [10,11,12,13,14,15,16,17,18]. However, there are only very few long-term follow-up studies investigating more than two biomarkers at multiple time points within the same patient cohort and not all biomarkers were available for all subjects in these studies [19,20,21]. To our knowledge, no previous study investigated effect sizes and minimal samples sizes to detect changes of these biomarkers within one monocentric cohort over time.
In this long-term predefined 24-month follow-up study we set out to determine effect sizes and minimal sample sizes for three imaging biomarkers in the same monocentric Alzheimer’s disease cohort: Aβ deposition measured with PiB-PET, neuronal dysfunction measured with FDG-PET, and neuronal loss measured by cortical thickness on MRI.
Methods
Ethics statement
The study protocol was approved by the German radiation protection authority and the ethics committee of the School of Medicine of the Technical University of Munich, Munich, Germany (reference number 1285/05). All patients provided written informed consent prior to any study-specific procedures and all clinical investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Patient recruitment and study design
Patients were recruited from the outpatient clinic of the Centre for Cognitive Disorders at the Department of Psychiatry, Klinikum rechts der Isar, Technical University of Munich, School of Medicine, Munich, Germany. They underwent a standardized diagnostic procedure including a neuropsychological evaluation as described previously [22]. This workup included an interview with the patient and an informant, obtaining demographic data, medical history, and concomitant medication as well as physical, neurological, and psychiatric examinations, a neuropsychological evaluation including the Mini-Mental State Examination (MMSE) [23], the Consortium to Establish a Registry for Alzheimer’s Disease Neuropsychological Assessment Battery (CERAD-NAB) [24], a routine laboratory screening test, and Apolipoprotein E genotyping [25]. The severity of cognitive impairment was rated on the Clinical Dementia Rating scale sum of boxes (CDR-SOB) [26]. It is calculated by adding the ratings of the individual domains of the CDR, resulting in a sum-score between 0 and 18. This allows for a finer gradation and monitoring of symptom progression than using the CDR-global score which gives information about the over-all severity of dementia on a scale between 0 and 3 [27, 28]. Accordingly, while changes in individual categories increase the CDR-SOB score the CDR-global score may remain unchanged. As a consequence, associations between clinical worsening of dementia and changes of individual biomarkers might be missed.
Imaging procedures included cranial MRI to assess structural brain abnormalities and cortical thickness, cerebral FDG-PET to determine neuronal metabolic deficit, and cerebral PiB-PET to assess brain amyloid deposition. All baseline assessments were completed within 1 month for each subject. Follow-up assessments were planned to take place 24 months after the initial evaluation.
In order to participate in this study, subjects needed to have AD typical hypometabolism on FDG-PET [29], as well as positive PiB uptake on visual analysis of the scans. Scans were evaluated by an experienced nuclear medicine specialist. All study participants met the National Institute on Aging and Alzheimer’s Association (NIA-AA) diagnostic criteria for early Alzheimer’s disease [6, 30], and can be placed in the Alzheimer’s continuum (A + T*(N)+: evidence of abnormal biomarkers for Aβ and neuronal injury; a biomarker for tau is not available as indicated by the asterisk) [31]. CDR-sum of boxes (SOB) scores of the subjects ranged between 0.5–9.0.
Exclusion criteria were described previously [22] and included other neurologic or psychiatric disorders, major morphologic or vascular MRI abnormalities, and patients with other possible causes of cognitive impairment such as psychotropic medication or major abnormalities in routine blood testing.
Brain imaging
Patients underwent cranial MRI examinations on a Siemens 1.5 Tesla Magnetom Symphony scanner using a standardized imaging protocol which consisted of a 3D T1 dataset (TR 1520 ms, TE 3.93 ms, 256 × 256 matrix, flip angle 15°, 1 mm slices), axial T2 weighted turbo-spin-echo images (TR 4510 ms, TE 104 ms, 19 slices, voxel dimensions 0.6 mm × 0.5 mm × 6.0 mm), coronal T1 weighted spin echo images (TR 527 ms, TE 17 ms, 19 slices, voxel dimensions 0.9 mm × 0.9 mm × 6.0 mm), T2 weighted gradient echo images (TR 725, TE 29, 19 slices, voxel dimensions 0.7 mm × 0.7 mm × 6.0 mm), and axial FLAIR images (TR 9000 ms, TE 105 ms, TI 2500 ms, 3 mm slices). Datasets were normalized to the MRI MNI-template in SPM8 to collect warping parameters for PET images. FDG-PET images were obtained using a Siemens ECAT HR+ PET scanner (CTI, Knoxville, Tenn., USA). Subjects received 370 MBq FDG at rest with eyes closed. Patients were positioned with the head parallel to the canthomeatal line within the gantry. Thirty minutes after injection, PET imaging was performed under standard resting conditions (eyes closed in dimmed ambient light). A sequence of one frame of 10 min and two frames of 5 min was started and later summed into a single frame. Image data were acquired in 3D mode with a total axial field of view of 15.5 cm. A transmission scan was acquired after completion of the emission scan for attenuation correction [32]. PiB-PET examinations were performed on the same scanner and followed a standardized protocol [33]. All patients were injected with 370 MBq PiB at rest outside the scanner. 30 min later they were placed in the scanner. At 40 mininutes post-injection, three 10-min frames of data acquisition were started and later summed into a single frame (40–70 min). Acquisition was carried out in 3D mode, and a transmission scan was carried out to allow for later attenuation correction.
The PiB and the FDG images were co-registered and normalized to the MNI space using the warping parameters of the MRI and smoothed using a Gaussian kernel of 10 mm × 10 mm × 10 mm [34]. PiB-PET, FDG-PET, and MRI of the brain were processed according to previously described standardized protocols using statistic parametric mapping software 8 (SPM 8 Wellcome Department of Cognitive Neurology, London, UK) in MATLAB 12 (The MathWorks, Inc., Natick, Massachusetts, USA) and FreeSurfer software Stable release version 5.1.0 [10, 14, 32, 34,35,36,37] (http://surfer.nmr.mhg.harvard.ed/; http://www.freesurfer.net/fswiki/VolmeRoiCorticalThickness).
Statistical analyses
To control for between-subjects differences in PiB uptake, standardized uptake value ratios (SUVRs) were obtained by calculating the cerebral to cerebellar vermis (C/cv) ratio for each patient as demonstrated previously [10, 34]. SUVRs of FDG uptake in AD signature regions [38] were similarly obtained with the exception that the pons was used as reference region (C/pons) [10]. The anatomical ROIs were defined using an established template [37]. For both PET modalities mean standard uptake volume values were calculated for 100% of voxels for each ROI.
Global cortical thickness score per participant was calculated using the averaged cortical thickness values as defined in the Desikan-Killiany-Tourville (DKT) protocol [39] in FreeSurfer software as described previously [36].
Differences between baseline and follow-up examination for all patients, irrespectively of the clinical stage within the AD continuum, were calculated for PiB uptake, FDG uptake, cortical thickness, CDR-SOB and MMSE and adjusted to a 24 months follow-up period.
Based on mean values and standard deviations post-hoc effect sizes (Cohen’s d) and minimal sample sizes (n) to detect a statistically significant change (two-tailed test, α = 5%, β = 20%) of each biomarker were calculated using G*power 3.1 [40, 41].
Results
Patients
The early AD patient sample is described in Table 1.
Effect sizes of imaging biomarkers and minimal sample sizes
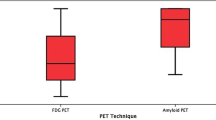
Post hoc power calculation for PiB-PET, FDG-PET, and cortical thickness revealed effect sizes of Cohens’s d = 1.315, 0.341, and 0.914, respectively. Accordingly, sample sizes of at least n = 7 for PiB-PET, n = 70 for FDG-PET, and n = 12 for cortical thickness are required to detect statistically significant changes of AD imaging biomarkers over time with a power of 80% (α = 5%, β = 20%). Results of global imaging data and power analyses for effect sizes and minimum sample sizes are shown in Table 2 and Fig. 1.

Effect sizes and minimal sample sizes based on post-hoc power calculations for the individual biomarkers to monitor disease progression in early AD in a prospective 24-month follow-up study. n: number; FDG-PET: 18F-fluordeoxyglucose positron emission tomography; PiB-PET: 11C-Pittsburgh compound B (PiB)
Discussion
Post hoc power calculation revealed large effect sizes for PiB-PET and cortical thickness, and a smaller effect size for FDG-PET with regards to sensitivity to pick up AD progression over a pre-defined follow-up interval of 24 months in an early AD sample. Although true effect sizes tend to be overestimated in smaller samples and therefore minimum sample sizes would probably need to be larger than estimated based on our results, amyloid PET and cortical thickness require markedly smaller sample sizes compared to FDG-PET to monitor AD disease progression with surrogate biomarkers.
The strengths of our study include long-term follow-up data of three imaging biomarkers measured in parallel within a monocentric sample. Follow-up occurred after a predefined interval of 24 months. We specifically defined inclusion and exclusion criteria, biomarkers, and the interval between baseline and follow-up to match trials investigating disease modifying drugs in early AD. Hence, our results might contribute to selecting biomarkers and estimate sample sizes for tracking pathognomonic AD changes by surrogate biomarkers in future AD trials.
The strength of a monocentric sample might also be conceived as a limitation as clinical trials are multicentric. The biggest limitation, however, is the sample size. While the number of participants is sufficient for the applied statistics, results would be more robust using larger sample sizes from multiple centres. Although we aimed for the follow-up examination to take place after 24 months in some cases this was not obtainable. We adjusted for time deviations assuming a linear change which could be debated [42]. According to the model of Jack et al. biomarkers in AD change in a specific order over the course of the disease [42]. In this study, different measures typically showing more or less dynamic changes during the stage of early AD were assessed. At this point, it is important to keep in mind that we did not set out to compare how well suited the individual biomarkers are to support the diagnosis of AD pathology rather than to investigate how well they are suited to show progression in an early AD cohort such as it would be recruited for a clinical trial. In addition, one could argue that there might be better ways to analyse the imaging measures. Chen et al. and Su et al. both saw significantly greater power to detect Aβ increase on amyloid PET after 24 months when using cerebral-to white matter SUVR changes instead of using the cerebellum as reference region [18, 43]. Also other factors influencing the rate of amyloid accumulation such as white matter hyperintensities [44], baseline SUVR [45], and the status of markers for amyloid and neuronal injury at baseline [21] would have to be taken into account when planning biomarker assessments for a clinical trial. Chen et al. calculated minimal sample sizes for an assumed 12-month clinical trial. Comparable to the results of our study samples of n = 8 for Aβ positive subjects with probable Alzheimer’s dementia and n = 13 for Aβ positive subjects with MCI would be necessary to detect a 25% decrease of Aβ SUVR [18]. Considering the high sensitivity and specificity of FDG-PET to detect AD [46] we were surprised by its comparably small effect size regarding monitoring of disease progression. A possible explanation could be the method of evaluating FDG uptake. In contrast to amyloid PET where there is no or minimal specific tracer uptake in the reference region SUVRs for FDG-PET are obtained using a reference region with specific tracer uptake. Although reference regions chosen for FDG-PET are thought to be less affected by Alzheimer’s disease, in a complex network system like the brain, neuronal activity in one region might affect neuronal activity in other regions and thus could possibly affect uptake in the reference region and consequently SUVRs between baseline and follow-up. While results from dynamic tracer imaging might be more robust acquiring this data is more complex and necessitates invasive procedures such as arterial blood lines. Under the consideration of patients’ safety and feasibility it might not be a suitable approach during a clinical trial.
Lastly, the selection of and the confinement to imaging markers could be debated. Despite already comparing three different biomarkers in the same highly characterised cohort, other biomarkers such as tau-PET, CSF or blood based biomarkers might be even more powerful to monitor disease progression. Although we did not investigate any of the 18F-labeled tracers for Aβ it can be reasoned that results would be similar to the results we described for 11C labelled PiB-PET. However, this would have to be further investigated.
Conclusion
When considering study designs amyloid imaging with PET and measuring cortical thickness with MRI are powerful biomarkers requiring relatively small sample sizes to monitor disease progression in early AD.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. However, due to the nature of pseudonymized patient data, a material transfer agreement is required to meet ethical standards and data privacy laws of Germany.
Change history
05 March 2020
Following publication of the original article [1], the authors ask to correct the surname of co-author Dennis Hedderich from from Heddderich to Hedderich.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADAS-cog:
-
Alzheimer’s disease assessment scale – cognitive subscale
- Aβ:
-
Amyloid beta
- C:
-
Cerebrum
- CDR-SOB:
-
Clinical dementia rating scale sum of boxes
- CSF:
-
Cerebro spinal fluid
- cv:
-
Cerebellar vermis
- FDG:
-
18F-fluordeoxyglucose
- MCI:
-
Mild cognitive impairment
- MRI:
-
Magnetic resonance imaging
- PET:
-
Positron emission tomography
- PiB:
-
11C-Pittsburgh compound B
- ROI:
-
Region of interest
- SUVR:
-
Standardized uptake value ratio
References
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–64.
Morris JC, Ernesto C, Schafer K, Coats M, Leon S, Sano M, Thal LJ, Woodbury P. Clinical dementia rating training and reliability in multicenter studies: the Alzheimer’s disease cooperative study experience. Neurology. 1997;48:1508–10.
Eli Lilly and Company, Progress of mild Alzheimer’s disease in participants on solanezumab versus placebo (EXPEDITION 3), 2013, ClinicalTrials Solanezumab.
Eli Lilly and Company, An efficacy and safety study of Lanabecestat (LY3314814) in early Alzheimer’s disease (AMARANTH), 2014.
Hoffmann-La R. CREAD study: a study of crenezumab versus placebo to evaluate the efficacy and safety in participants with prodromal to mild Alzheimer’s disease (AD); 2016.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012;11:1006–12.
Podhorna J, Krahnke T, Shear M, Harrison JE, I. Alzheimer’s Disease Neuroimaging. Alzheimer’s disease assessment scale-cognitive subscale variants in mild cognitive impairment and mild Alzheimer’s disease: change over time and the effect of enrichment strategies. Alzheimers Res Ther. 2016;8:8.
Morris JC, Selkoe DJ. Recommendations for the incorporation of biomarkers into Alzheimer clinical trials: an overview. Neurobiol Aging. 2011;32(Suppl 1):S1–3.
Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry. 2009;65:927–34.
Jack CR Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–80.
Habeck C, Foster NL, Perneczky R, Kurz A, Alexopoulos P, Koeppe RA, Drzezga A, Stern Y. Multivariate and univariate neuroimaging biomarkers of Alzheimer’s disease. Neuroimage. 2008;40:1503–15.
Adriaanse SM, van Dijk KR, Ossenkoppele R, Reuter M, Tolboom N, Zwan MD, Yaqub M, Boellaard R, Windhorst AD, van der Flier WM, Scheltens P, Lammertsma AA, Barkhof F, van Berckel BN. The effect of amyloid pathology and glucose metabolism on cortical volume loss over time in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41:1190–8.
Grimmer T, Henriksen G, Wester HJ, Forstl H, Klunk WE, Mathis CA, Kurz A, Drzezga A. Clinical severity of Alzheimer’s disease is associated with PIB uptake in PET. Neurobiol Aging. 2009;30:1902–9.
Klupp E, Grimmer T, Tahmasian M, Sorg C, Yakushev I, Yousefi BH, Drzezga A, Forster S. Prefrontal hypometabolism in Alzheimer disease is related to longitudinal amyloid accumulation in remote brain regions. J Nucl Med. 2015;56:399–404.
Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008;4:38–48.
Morris E, Chalkidou A, Hammers A, Peacock J, Summers J, Keevil S. Diagnostic accuracy of (18) F amyloid PET tracers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging. 2016;43:374–85.
Chen K, Roontiva A, Thiyyagura P, Lee W, Liu X, Ayutyanont N, Protas H, Luo JL, Bauer R, Reschke C, Bandy D, Koeppe RA, Fleisher AS, Caselli RJ, Landau S, Jagust WJ, Weiner MW, Reiman EM, I. Alzheimer’s Disease Neuroimaging. Improved power for characterizing longitudinal amyloid-beta PET changes and evaluating amyloid-modifying treatments with a cerebral white matter reference region. J Nucl Med. 2015;56:560–6.
Beckett LA, Harvey DJ, Gamst A, Donohue M, Kornak J, Zhang H, Kuo JH, I. Alzheimer’s Disease Neuroimaging. The Alzheimer’s disease neuroimaging initiative: annual change in biomarkers and clinical outcomes. Alzheimers Dement. 2010;6:257–64.
Gomar JJ, Conejero-Goldberg C, Davies P, Goldberg TE, I. Alzheimer’s Disease Neuroimaging. Extension and refinement of the predictive value of different classes of markers in ADNI: four-year follow-up data. Alzheimers Dement. 2014;10:704–12.
Bertens D, Tijms BM, Vermunt L, Prins ND, Scheltens P, Visser PJ. The effect of diagnostic criteria on outcome measures in preclinical and prodromal Alzheimer’s disease: implications for trial design. Alzheimers Dement (N Y). 2017;3:513–23.
Ortner M, Kurz A, Alexopoulos P, Auer F, Diehl-Schmid J, Drzezga A, Forster S, Forstl H, Perneczky R, Sorg C, Yousefi BH, Grimmer T. Small vessel disease, but neither amyloid load nor metabolic deficit, is dependent on age at onset in Alzheimer's disease. Biol Psychiatry. 2015;77:704–10.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, Mellits ED, Clark C. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology. 1989;39:1159–65.
Zivelin A, Rosenberg N, Peretz H, Amit Y, Kornbrot N, Seligsohn U. Improved method for genotyping apolipoprotein E polymorphisms by a PCR-based assay simultaneously utilizing two distinct restriction enzymes. Clin Chem. 1997;43:1657–9.
Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(Suppl 1):173–6 discussion 177-8.
Lynch CA, Walsh C, Blanco A, Moran M, Coen RF, Walsh JB, Lawlor BA. The clinical dementia rating sum of box score in mild dementia. Dement Geriatr Cogn Disord. 2006;21:40–3.
O'Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ, Lupo PJ, Reisch JS, Doody R, C. Texas Alzheimer’s Research. Staging dementia using clinical dementia rating scale sum of boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol. 2008;65:1091–5.
Minoshima S. Imaging Alzheimer’s disease: clinical applications. Neuroimaging Clin N Am. 2003;13:769–80.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, Contributors. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–62.
Grimmer T, Diehl J, Drzezga A, Forstl H, Kurz A. Region-specific decline of cerebral glucose metabolism in patients with frontotemporal dementia: a prospective 18F-FDG-PET study. Dement Geriatr Cogn Disord. 2004;18:32–6.
Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, Meltzer CC, Schimmel K, Tsopelas ND, DeKosky ST, Price JC. Simplified quantification of Pittsburgh compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–72.
Drzezga A, Grimmer T, Henriksen G, Stangier I, Perneczky R, Diehl-Schmid J, Mathis CA, Klunk WE, Price J, DeKosky S, Wester HJ, Schwaiger M, Kurz A. Imaging of amyloid plaques and cerebral glucose metabolism in semantic dementia and Alzheimer's disease. Neuroimage. 2008;39:619–33.
Mazziotta JC, Toga AW, Evans A, Fox P, Lancaster J. A probabilistic atlas of the human brain: theory and rationale for its development. The International Consortium for Brain Mapping (ICBM). Neuroimage. 1995;2:89–101.
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–5.
Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–89.
Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ, I. Alzheimer’s Disease Neuroimaging. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207–18.
Klein A, Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front Neurosci. 2012;6:171.
Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41:1149–60.
Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91.
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–16.
Su Y, Blazey TM, Owen CJ, Christensen JJ, Friedrichsen K, Joseph-Mathurin N, Wang Q, Hornbeck RC, Ances BM, Snyder AZ, Cash LA, Koeppe RA, Klunk WE, Galasko D, Brickman AM, McDade E, Ringman JM, Thompson PM, Saykin AJ, Ghetti B, Sperling RA, Johnson KA, Salloway SP, Schofield PR, Masters CL, Villemagne VL, Fox NC, Forster S, Chen K, Reiman EM, Xiong C, Marcus DS, Weiner MW, Morris JC, Bateman RJ, Benzinger TL, N. Dominantly Inherited Alzheimer. Quantitative amyloid imaging in autosomal dominant Alzheimer’s disease: results from the DIAN study group. PLoS One. 2016;11:e0152082.
Grimmer T, Faust M, Auer F, Alexopoulos P, Forstl H, Henriksen G, Perneczky R, Sorg C, Yousefi BH, Drzezga A, Kurz A. White matter hyperintensities predict amyloid increase in Alzheimer’s disease. Neurobiol Aging. 2012;33:2766–73.
Guo T, Dukart J, Brendel M, Rominger A, Grimmer T, Yakushev I, I. Alzheimer’s Disease Neuroimaging. Rate of beta-amyloid accumulation varies with baseline amyloid burden: implications for anti-amyloid drug trials. Alzheimers Dement. 2018;14:1387–96.
Bloudek LM, Spackman DE, Blankenburg M, Sullivan SD. Review and meta-analysis of biomarkers and diagnostic imaging in Alzheimer’s disease. J Alzheimers Dis. 2011;26:627–45.
Acknowledgements
Not applicable.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MO: Acquisition and analysis of data, drafting a significant portion of the manuscript; RD: Acquisition and analysis of data, drafting a significant portion of the manuscript, DH: Acquisition and analysis of data; OG: Acquisition and analysis of data, FMS: Acquisition and analysis of data; JDS: Acquisition and analysis of data, drafting a significant portion of the manuscript; HF: drafting a significant portion of the manuscript; IY: Acquisition and analysis of data, TG: Conception and design of the study; acquisition and analysis of data, drafting a significant portion of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the German radiation protection authority and the ethics committee of the Faculty of Medicine of the Technical University of Munich, Munich, Germany (reference number 1285/05). All patients provided written informed consent prior to any study specific procedures and all clinical investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ortner, M., Drost, R., Heddderich, D. et al. Amyloid PET, FDG-PET or MRI? - the power of different imaging biomarkers to detect progression of early Alzheimer’s disease. BMC Neurol 19, 264 (2019). https://doi.org/10.1186/s12883-019-1498-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-019-1498-9