
Abstract
Background
Neuromyelitis optica (NMO) is a severely disabling inflammatory disorder of the central nervous system and is often misdiagnosed as multiple sclerosis (MS). There is increasing evidence that treatment options shown to be beneficial in MS, including interferon-β (IFN-β), are detrimental in NMO.
Case presentation
We here report the first Caucasian patient with aquaporin 4 (AQP4) antibody (NMO-IgG)-seropositive NMO presenting with a tumefactive brain lesion on treatment with IFN-β. Disease started with relapsing optic neuritis and an episode of longitudinally extensive transverse myelitis (LETM) in the absence of any brain MRI lesions or cerebrospinal fluid-restricted oligoclonal bands. After initial misdiagnosis of multiple sclerosis (MS) the patient received subcutaneous IFN-β1b and, subsequently, subcutaneous IFN-β1a therapy for several years. Under this treatment, the patient showed persisting relapse activity and finally presented with a severe episode of subacute aphasia and right-sided hemiparesis due to a large T2 hyperintensive tumefactive lesion of the left brain hemisphere and a smaller T2 lesion on the right side. Despite rituximab therapy two further LETM episodes occurred, resulting in severe neurological deficits. Therapeutic blockade of the interleukin (IL)-6 signalling pathway by tocilizumab was initiated, followed by clinical and radiological stabilization.
Conclusion
Our case (i) illustrates the relevance of correctly distinguishing NMO and MS since these disorders differ markedly in their responsiveness to immunomodulatory and -suppressive therapies; (ii) confirms and extends a previous report describing the development of tumefactive brain lesions under IFN-β therapy in two Asian NMO patients; and (iii) suggests tocilizumab as a promising therapeutic alternative in highly active NMO disease courses.
Similar content being viewed by others

Background
Interferon-β (IFN-β) is one of the established first-line therapies for patients with relapsing-remitting multiple sclerosis (MS). While the efficacy of IFN-β in MS is widely accepted, the possible benefit in non-classical MS variants has been disputed. This is the case for neuromyelitis optica (NMO/Devic’s syndrome), considered an MS subtype until the discovery of the anti-aquaporin 4 (AQP4) antibodies (AQP4-Ab) [1],[2]. In both Asian and European NMO patients, IFN-β failed to show therapeutic efficacy [3]-[5]. Of note, Shimizu and colleagues recently reported two Asian patients with NMO spectrum disease (NMOSD) who developed extensive brain lesions following IFN-β therapy [6]. Here we report the case of an AQP4 Ab-seropositive NMO patient of Caucasian descent presenting with a tumefactive lesion on IFN-β treatment. Of note, our patient did not respond to B cell depletion by the monoclonal anti-CD20 antibody rituximab but improved upon blockade of the interleukin (IL)-6 pathway using tocilizumab.
Case Presentation
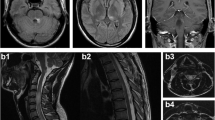
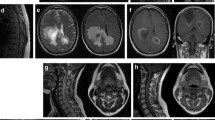
A 47-year old Caucasian female patient had been diagnosed with MS in 1995 in an external hospital after four episodes of optic neuritis (ON) since 1989 and an episode of longitudinally extensive transverse myelitis (LETM) in 1995 (Figure 1). Cerebrospinal fluid (CSF)-restricted oligoclonal bands (OCBs) were repeatedly negative and brain MRI scans did not show any abnormalities (e.g. MRI performed in 2001, Figure 1). After an initial two months azathioprine treatment in 1995, which had to be discontinued because of side effects, repeated monthly intravenous (iv) steroid pulse treatments with 250 mg methylprednisolone (MP) were initiated and continued until April 2013. In 2001, after three further episodes of ON, subcutaneous IFN-β1b medication was added to the ongoing steroid pulse treatment. In 2005, subcutaneous IFN-β1b was switched to subcutaneous IFN-β1a (3 × 44 μg/week). During IFN-β therapy the patient still suffered from further relapses, most of them occurring between the years 2001 and 2005 (July 2002, February 2005, and April 2005). She recovered only partially from the relapses and accumulated fixed neurological deficits such as amaurosis of the right eye and impaired walking with an expanded disability status scale (EDSS) score of 4.0 in January 2012. Of note, there was no chronic progression between the relapses. In 2011 persisting disease activity prompted testing for serum AQP4 antibodies (by a cell-based assay; CBA [7]) which were found positive and confirmed the diagnosis of seropositive NMO. Following the patient’s wishes and in light of the reduced relapse rate since 2005 therapy with subcutaneous IFN-β1a was continued, together with the regular monthly steroid therapy (250 mg MP each) established since 1995. In January 2012, she suffered from a subacute aphasia and right sided hemiparesis and presented to our department. MRI imaging showed a large T2 hyperintensive, tumefactive lesion in the prefrontal white matter of the superior and middle frontal gyrus of the left hemisphere and smaller T2 lesions on the right side without clear evidence of a disturbed blood brain barrier (Figure 1). MR spectroscopy showed an unchanged choline peak and thus did not give a hint to a malignant process. Analysis of CSF was unremarkable with still no quantitative evidence for intrathecal immunoglobulin (Ig) synthesis or OCBs and a normal CSF white cell count. AQP4-Ab serum titre was 1:1000 (CBA). Furthermore, a time-of-flight MR-angiography showed no signs of vasculitis, and laboratory work-up revealed no clinical or serological evidence for connective tissue diseases. The patient received a high-dose methylprednisolone iv pulse and clinically improved. The follow-up MRI one month later showed a mild regression of the large T2 hyperintense lesion. We stopped IFN-β therapy and continued the already existing monthly steroid pulses (250 mg MP each) as the patient declined any additional medication at that time. However, two attacks of myelitis occurred in March 2013 and April 2013. Follow-up AQP4-Ab testing in April 2013 confirmed seropositivity (1:1000; CBA). The patient was treated with high-dose steroid iv pulse therapy and rescue plasmapheresis. Thereafter our patient agreed to an adaptation of therapy and the monthly steroid infusions were discontinued without tapering.

Disease course, MRI scans and therapy in a patient with AQP4-Ab-positive NMO. The figure depicts clinical relapses (diamonds), MRI findings (T2, FLAIR), and therapy data (methyl prednisolone (MP) 1000 mg i.v. 3–5 daily infusions; tocilizumab 8 mg/kg bodyweight once a month; rituximab 1000 mg i.v. (single infusion), IFN (interferon)-β1a 44 μg s.c. three times a week (Rebif®); IFN-β1b s.c. daily (Betaferon®, Betaseron®); methylpredisolone 250 mg i.v. once per month; azathioprine (AZA)). No relapses have occurred during therapy with tocilizumab (follow-up: 12 months). AQP: aquaporin, AZA: azathioprine, FLAIR: fluid attenuated inversion recovery, IFN: interferon, MP: methyl prednisolone, MRI: magnetic resonance imaging, MS: multiple sclerosis, NMO: neuromyelitis optica.
We initiated treatment with rituximab (single intravenous infusion of 1000 mg) in May 2013 and transferred our patient to a rehabilitation centre. However, further severe LETM attacks occurred shortly thereafter in June and August 2013. The patient was re-admitted to our department in September 2013 and showed persistently increased AQP4-Ab titres (1:3200, CBA). Absolute B cell counts were normal (68/μl) while relative proportion was decreased (3% of all lymphocytes; reference value 5-24%). This relapse was associated with a urinary tract infection (without systemic signs of inflammation, i.e. without leucocytosis, fever or elevated C-reactive protein (CRP) levels) which we successfully treated according to local guidelines. In light of the patient’s severe clinical deficits (EDSS of 9.0) we applied another iv steroid pulse and decided to switch to tocilizumab, a humanized monoclonal anti-IL-6-receptor antibody. Monthly infusions of tocilizumab with 8 mg/kg bodyweight were followed by clinical stabilization as no further relapses occurred for the following 12 months (see clinical and MRI follow-up after withdrawal of IFN-β in Figure 1). We also observed a slight clinical improvement, as reflected by an EDSS of 8.0. Of note, serum AQP4 antibodies were still detectable at high titre at last follow-up in October 2014 (1:3200, CBA).
Conclusions
To our knowledge, we here present the first Caucasian NMO patient who developed a tumefactive brain lesion on long-term IFN-β treatment. Our case confirms and extends similar findings in two Asian NMOSD patients recently reported by Shimizu and colleagues [6]. Obviously the development of a tumefactive brain lesion during IFN-β therapy of NMO is not linked to a specific ethnicity. Moreover, in line with results obtained previously in other cohorts, our patient experienced numerous relapses under IFN-β therapy. Why NMO patients fail to respond to IFN-β remains so far unclear. Possible explanations consider the complex immunomodulatory effects of IFN-β, including the upregulation of proinflammatory cytokines observed in MS [8]. Since in NMOSD higher CNS levels of IL-17 are observed than in MS [9], it may be speculated that IL-17 biology may play a role in NMO unresponsiveness to IFN-β. Moreover, a recent study in NMO patients could link serum levels of IFN-α, a type I interferon closely related to IFN-β, with clinical disease activity and severity [10]. Independent experimental work demonstrated that NMO-IgG-mediated astrocyte damage is reduced in the CNS of type I interferon receptor deficient mice, suggesting that type I interferon signalling contributes to the pathogenesis of neuromyelitis optica [11].
Of note, administration of a single intravenous infusion of 1000 mg rituximab was followed by further relapses just one and three months later. While CD19 counts were not determined directly after rituximab application, an investigation four months after rituximab dosing showed normal absolute B cell counts and persistently increased AQP4-Ab levels. We may speculate that our patient belongs to individuals characterized by an early repopulation of the B cell compartment following rituximab therapy [12].
Our case adds preliminary evidence in favour of a possible beneficial effect of tocilizumab in NMO, even following treatment with rituximab [13]-[15]. However, considering that NMO is often characterized by very long periods free of any relapses, the suggested success of tocilizumab in our patient needs to be followed up. Moreover, more studies, in particular independent controlled trials, are required to confirm the efficacy of tocilizumab in NMO suggested by this case report and previous ones [13]-[16]. The demonstration of significantly increased levels of IL-6 in comparison to MS in the CSF and serum of patients with NMO [17],[18], particularly in AQP4 Ab-seropositive patients with severe disease activity [18], provides a rationale for the use of this drug in NMO. In an ex vivo experimental model of NMO spinal cord slice cultures exposed to NMO-IgG and complement showed a marked loss of AQP4 and myelin which was enhanced by adding IL-6 [19]. Moreover, Chihara and colleagues described a specific IL-6-dependent B lymphocyte subpopulation in the peripheral blood and CSF of NMO patients: these CD19+ CD27+ CD38+ CD180+ B cells were found to produce AQP4 antibodies and showed enhanced survival as well as AQP4 antibody secretion in the presence of IL-6, whereas blockage of the IL-6 receptor signalling by an anti-IL-6R antibody shortened their survival in vitro [20].
Our report illustrates the importance of correctly diagnosing NMO and MS to avoid mistreatment with potentially severe or even fatal consequences. Two recent studies have revealed that up to 30% of patients with NMO were falsely diagnosed with MS [21],[22] and, in consequence, often treated with drugs not effective in NMO. While AQP4-IgG serum testing is certainly the most important differential diagnostic measure, other investigations such as lumbar puncture or brain and spinal cord MRI are important as well. In the present case, the diagnosis of MS was made in the absence of CSF-restricted OCBs. OCB negativity is extremely rare in MS (~2-5%) and should prompt physicians to question that diagnosis; by contrast, around 70% of patients with AQP4-IgG-positive NMOSD lack OCBs [22]-[24]. Normal CSF/serum concentration quotient values of IgG (QIgG) in NMOSD are especially common during periods of clinical remission [23]. Similarly, a persistently normal brain MRI as observed in our patient should warn against the diagnosis of MS [25]. Brain lesions are absent in most patients with NMO at disease onset, but may occur later in the disease and may even meet MRI criteria for MS [23],[26]. Of note, for Asian NMO patients, independent case reports described the occurrence of hemispheric brain lesions even without association to IFN-β therapy [27]-[29], while we are not aware of such observations for NMO patients of other ethnic background. Finally, predominantly central, longitudinally extensive spinal cord lesions extending over more than three vertebral segments are extremely rare in MS while their presence is highly suggestive of NMOSD [30].
Our case supports the conclusion that the development of tumefactive brain lesions under IFN-β therapy for suspected MS should prompt considering NMOSD as the underlying disease, obviously not only in Asian but also in Caucasian patients. Moreover, our report underlines – in accordance with previous reports and recommendations [31] – the need to treat MS and NMO, two conditions with substantial differences in pathogenesis [32],[33] differentially and, in particular, to avoid treatment with IFN-β in patients with NMO. Our report adds to previous evidence indicating a potential treatment effect of tocilizumab, an IL-6 signalling pathway blocker in NMO therapy.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
References
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR: IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005, 202: 473-477. 10.1084/jem.20050304.
Jarius S, Wildemann B: The history of neuromyelitis optica. J Neuroinflamm. 2013, 10: 8-10.1186/1742-2094-10-8.
Uzawa A, Mori M, Hayakawa S, Masuda S, Kuwabara S: Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur J Neurol. 2010, 17: 672-676. 10.1111/j.1468-1331.2009.02897.x.
Palace J, Leite MI, Nairne A, Vincent A: Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010, 67: 1016-1017. 10.1001/archneurol.2010.188.
Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H, Goto J, Shimizu T, Takatsu M, Sakurai Y, Nakase H, Uesaka Y, Hashida H, Hashimoto K, Komiya T, Tsuji S: IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010, 75: 1423-1427. 10.1212/WNL.0b013e3181f8832e.
Shimizu Y, Yokoyama K, Misu T, Takahashi T, Fujihara K, Kikuchi S, Itoyama Y, Iwata M: Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J Neurol. 2008, 255: 305-307. 10.1007/s00415-007-0730-5.
Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, Wandinger KP: Standardized method for the detection of antibodies to aquaporin-4 based on a highly sensitive immunofluorescence assay employing recombinant target antigen. J Neurol Sci. 2010, 291: 52-56. 10.1016/j.jns.2010.01.002.
Wandinger K, Stürzebecher C, Bielekova B, Detore G, Rosenwald A, Staudt LM, McFarland HF, Martin R: Complex immunomodulatory effects of interferon-? in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Ann Neurol. 2001, 50: 349-357. 10.1002/ana.1096.
Ishizu T, Osoegawa M, Mei F, Kikuchi H, Tanaka M, Takakura Y, Minohara M, Murai H, Mihara F, Taniwaki T, Kira J: Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 2005, 128: 988-1002. 10.1093/brain/awh453.
Asgari N, Voss A, Steenstrup T, Kyvik KO, Stenager E, Lillevang ST: Interferon alpha association with neuromyelitis optica. Clin Dev Immunol. 2013, 2013: 713519-10.1155/2013/713519.
Khorooshi R, Wlodarczyk A, Asgari N, Owens T: Neuromyelitis optica-like pathology is dependent on type I interferon response. Exp Neurol. 2013, 247: 744-747. 10.1016/j.expneurol.2013.02.005.
Greenberg BM, Graves D, Remington G, Hardeman P, Mann M, Karandikar N, Stuve O, Monson N, Frohman E: Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler. 2012, 18: 1022-1026. 10.1177/1352458511432896.
Kieseier BC, Stüve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK, Ringelstein M, Turowski B, Aktas O, Antoch G, Hartung H: Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013, 70: 390-393. 10.1001/jamaneurol.2013.668.
Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, Gold R: Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013, 70: 394-397. 10.1001/jamaneurol.2013.1246.
Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, Miyake S, Aranami T, Yamamura T: Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014, 82: 1302-1306. 10.1212/WNL.0000000000000317.
Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T: Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol. 2013, 23: 827-831. 10.3109/s10165-012-0715-9.
Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S, Taniguchi J, Kuwabara S: Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler. 2010, 16: 1443-1452. 10.1177/1352458510379247.
Içöz S, Tüzün E, Kürtüncü M, Durmuş H, Mutlu M, Eraksoy M, Akman-Demir G: Enhanced IL-6 production in aquaporin-4 antibody positive neuromyelitis optica patients. Int J Neurosci. 2010, 120: 71-75. 10.3109/00207450903428970.
Zhang H, Bennett JL, Verkman AS: Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol. 2011, 70: 943-954. 10.1002/ana.22551.
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T: Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A. 2011, 108: 3701-3706. 10.1073/pnas.1017385108.
Mealy MA, Wingerchuk DM, Greenberg BM, Levy M: Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012, 69: 1176-1180.
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, et al: Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J Neuroinflamm. 2012, 9: 14-10.1186/1742-2094-9-14.
Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, Rommer P, Kleiter I, Stich O, Reuss R, Rauer S, Zettl UK, Wandinger KP, Melms A, Aktas O, Kristoferitsch W, Wildemann B: Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. 2011, 306: 82-90. 10.1016/j.jns.2011.03.038.
Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B: Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflamm. 2010, 7: 52-10.1186/1742-2094-7-52.
Traboulsee AL, Li DKB: The role of MRI in the diagnosis of multiple sclerosis. Adv Neurol. 2006, 98: 125-146.
Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG: Brain abnormalities in neuromyelitis optica. Arch Neurol. 2006, 63: 390-396. 10.1001/archneur.63.3.390.
Cheng C, Jiang Y, Chen X, Dai Y, Kang Z, Lu Z, Peng F, Hu X: Clinical, radiographic characteristics and immunomodulating changes in neuromyelitis optica with extensive brain lesions. BMC Neurol. 2013, 13: 72-10.1186/1471-2377-13-72.
Ikeda K, Ito H, Hidaka T, Takazawa T, Sekine T, Yoshii Y, Hirayama T, Kawabe K, Kano O, Iwasaki Y: Repeated non-enhancing tumefactive lesions in a patient with a neuromyelitis optica spectrum disorder. Intern Med. 2011, 50: 1061-1064. 10.2169/internalmedicine.50.4295.
Nakamura M, Endo M, Murakami K, Konno H, Fujihara K, Itoyama Y: An autopsied case of neuromyelitis optica with a large cavitary cerebral lesion. Mult Scler. 2005, 11: 735-738. 10.1191/1352458505ms1236cr.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG: The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6: 805-815. 10.1016/S1474-4422(07)70216-8.
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kümpfel T: Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. 2014, 261: 1-16. 10.1007/s00415-013-7169-7.
Jarius S, Wildemann B: AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010, 6: 383-392. 10.1038/nrneurol.2010.72.
Papadopoulos MC, Bennett JL, Verkman AS: Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014, 10: 493-506. 10.1038/nrneurol.2014.141.
Acknowledgements
We thank the patient for giving her consent to publishing this case report. We thank the Walter-und-Ilse-Rose-Stiftung for supporting the Multiple Sclerosis Center at the Department of Neurology, Heinrich-Heine University, Düsseldorf, Germany. S.J. thanks the Dietmar Hopp Foundation and Merck Serono for supporting research at the Department of Neurology, University of Heidelberg, Heidelberg, Germany. This work was supported by the Eugène Devic European Network (EDEN, BMBF 01GM1203A; EU-FP7).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
J.H. received speaker honoraria from Novartis and travel expenses from Merck Serono
M.R. received speaker honoraria from Novartis and travel reimbursement from Bayer Schering and Biogen Idec.
J.I. reports no conflicts of interest.
C.M. reports no conflicts of interest.
N.G. reports no conflicts of interest.
H.P.H. reports having received honoraria for consulting and speaking at symposia funded by Bayer Healthcare, Biogen Idec, GeNeuro, Genzyme, MedImmune, Merck Serono, Novartis, Roche, Teva, and Sanofi-Aventis, with approval by the Rector of Heinrich Heine University.
S.J. received travel compensation from the Guthy Jackson Charitable Foundation. The work of S.J. was directly supported by a research grant from the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) and indirectly by research grants from the Dietmar Hopp Stiftung, from Merck Serono and from Bayer Healthcare to the Division of Molecular Neuroimmunology, University of Heidelberg.
O.A. reports having received honoraria for consulting and speaking from Bayer HealthCare, Biogen Idec, Genzyme, Merck Serono, Novartis, Teva, Sanofi-Aventis, as well as research funding from the German Research Council (DFG), the Hertie Foundation and the Eugène Devic European Network (BMBF/EU-FP7).
Authors’ contributions
JH. acquired the data, drafted the manuscript and created the figure. MR. acquired the data and helped to draft the manuscript. JI. revised the manuscript for important intellectual content and helped creating the figure. CM. revised the manuscript for important intellectual content and helped creating the figure. NG revised the manuscript for important intellectual content. HPH revised the manuscript for important intellectual content. SJ revised the manuscript for important intellectual content. OA helped to draft the manuscript, revised the manuscript for important intellectual content and updated the figure. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Harmel, J., Ringelstein, M., Ingwersen, J. et al. Interferon-β-related tumefactive brain lesion in a Caucasian patient with neuromyelitis optica and clinical stabilization with tocilizumab. BMC Neurol 14, 247 (2014). https://doi.org/10.1186/s12883-014-0247-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-014-0247-3