
Abstract
Background
Hearing loss (HL) is the most common sensorineural disorder with high phenotypic and genotypic heterogeneity, which negatively affects life quality. Autosomal recessive non-syndromic hearing loss (ARNSHL) constitutes a major share of HL cases. In the present study, Whole exome sequencing (WES) was applied to investigate the underlying etiology of HL in an Iranian patient with ARNSHL.
Methods
A proband from an Iranian consanguineous family was examined via WES, following GJB2 sequencing. WES was utilized to find possible genetic etiology of the disease. Various Bioinformatics tools were used to assess the pathogenicity of the variants. Co-segregation analysis of the candidate variant was carried out. Interpretation of variants was performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines.
Results
WES results showed a novel frameshift (16 bp deletion) variant (p.Ala170Alafs*20) in the LRTOMT gene. This variant, which resides in exon 6, was found to be co-segregating in the family. It fulfils the criteria set by the ACMG guidelines of being pathogenic.
Conclusion
Here, we report successful application of WES to identify the molecular pathogenesis of ARNSHL, which is a genetically heterogeneous disorder, in a patient with ARNSHL.
Similar content being viewed by others


Background
Hearing loss (HL) is the most common congenital sensorineural defect affecting about 1 of 500–1000 newborns worldwide. It represents a significant global health problem [1]. HL has a wide spectrum of clinical manifestations: congenital or late onset, conductional or sensorineural, syndromic or non-syndromic [2]. Approximately, 50% of HL is related to genetic causes [2], meanwhile environmental and age-related HL account for the remaining percentage [3, 4]. Non-syndromic HL (NSHL) is responsible for 70–80% of all hereditary cases of HL. It, in turn, includes the autosomal recessive (AR) pattern (75%), the autosomal dominant (AD) (20–25%) and mitochondrial and X-linked HL (about 1%) [5,6,7]. Syndromic HL (SHL) accounts for the remaining 20–30% of genetic disorders in children [7]. There are about 400 types of syndromic HL [8]. HL is frequent in the Middle East and Northern African countries, with high rate of consanguinity [9, 10]. Iran, which has a consanguinity rate of about 38.6%, is an appropriate region for HL studies [11].
Mutations in one single locus, DFNB1 locus at 13q11–12 containing GJB2 (gap junction protein β-2) and GJB6 genes [7], account for 50% of the ARNSHL etiology in many Western populations [12]. In Iran, the prevalence of the GJB2 mutations is variable, depending on ethnicity and geographical location [13, 14]. The average percentage of GJB2 mutations, as the cause of ARNSHL, in Iran is about 18.7% [15], with a higher frequency in the north (33%) and a lower frequency (4%) in the southern regions. In this region, mutations in SLC26A4 are more frequent [16]. After the exclusion of GJB2, recognizing the underlying gene is difficult due to the high degree of genetic heterogeneity of HL. Therefore, whole exome sequencing would be ideal to determine HL causing mutations [17].
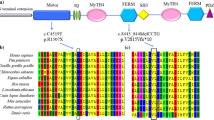
DFNB63 (OMIM 611451) was mapped to human chromosome 11q13.3-q13.4 [18,19,20]. This region contains the LRTOMT (Leucine Rich Transmembrane and O-Methyl-Transferase) gene [21]. LRTOMT is a fusion gene that has alternative reading frames and only exists in primates. Human LRTOMT has 10 exons, of which the first two are non-coding. Five transcripts have been reported for this gene. It has two different major protein products namely LRTOMT1 and LRTOMT2, which differ in the position of the start codons (Fig. 1a) [18]. LRTOMT is expressed in sensory hair cells with a fundamental role in auditory and vestibular functions [22]. LRTOMT1 is of unknown function while LRTOMT2 participates in inactivation of catecholamine neurotransmitters. Notably, there is homology between LRTOMT2 and COMT and the majority of the residues that are involved in the substrate binding region are conserved [23, 24]. Accordingly, it seems that LRTOMT2 might function as a catechol-O-methyltransferase [25]. Thus, LRTOMT2 has been named as COMT2. It catalyzes the transfer of a methyl group from S-adenosyl-L-methionine (AdoMet) to a hydroxyl group of catechols [23]. It is expressed in sensory hair cells in the inner ear. The defects in O-methyl transferase protein have been noted to cause NSHL [16]. Missense mutations cause a significant reduction in COMT2 enzymatic activity, suggesting that a defect in catecholamine catabolism underlies auditory and vestibular phenotypes [22]. The 11q13.3-q13.4 includes FGF3 (Fibroblast growth factor 3) gene, too. Mutations of this gene cause a form of syndromic HL (OMIM 610706), characterized by microtia, microdontia and inner ear agenesis [26, 27]. Patients with NSHL that are due to mutations in the LRTOMT gene have been found to be segregating only in the Middle Eastern, which is of a high consanguineous marriage [20, 21, 26, 28,29,30]. The highest mutation frequency in this gene is reported in Tunisian families and then in Iranian, Turkish and Pakistani families [21, 22]. These mutations lead to severe-to-profound prelingual NSHL [18].

a Two isoforms encoded by the LRTOMT gene: LRTOMT1 and LRTOMT2. LRTOMT1 starts in exon 3 and LRTOMT2 starts in exon 5. CDS regions are colored red. In this transcript of LRTOMT (NM_001145308), LRTOMT2, starts from exon 3 and ends in exon 7. b Pedigree of the family. The proband is marked by an arrow. c Pure tone audiogram of patient. Audiogram indicate sever-to-profound hearing loss in both ears. Frequency in hertz (Hz) and the hearing threshold in decibels (dB) are shown. d The electropherogram of the muatation in the patient (A1), deletion of 16 bp homozygously, in his father (A2) and his mother (A3) heterozygously. In the bottom of the electropherograms, the comparison of three sequence with refrence sequence is shown. The deleted 16 bp is shown in red box
The recently developed next generation sequencing (NGS) technologies such as targeted NGS (TNGS), whole genome sequencing (WGS) or whole exome sequencing (WES) have revolutionary improved disease diagnosis [31] and discovery of novel disease-causing genes and variants [32]. Recent advances in DNA enrichment and NGS methods have provided the opportunity for rapid and cost-effective analysis to identify pathogenic mutations in HL patients [33]. WES is an acceptable method with efficient strategy to recognize disease-causing mutations in genetically heterogeneous diseases. In this method, single nucleotide variants (SNVs), small insertion/deletions, and sometimes structural changes such as copy number variations (CNVs) can be diagnosed [34, 35].
The aim of this study was to identify the molecular pathology of congenital HL in a four-year-old boy using WES, which led to the identification of a novel frameshift mutation.
Methods
Subject and clinical evaluations
A four-year-old boy with congenital HL from an Iranian consanguineous family (first cousins) with no history of HL was ascertained (pedigree is shown in Fig. 1b). Comprehensive family history, audiological testing such as play audiometry, tympanometry, acoustic stapedial reflex, transient /distortion product oto acoustic emission (TE/DPOAEs), auditory brainstem response (ABR), auditory steady state response (ASSR) and physical examination were performed. The parents signed written informed consent following pre-test genetic counseling. Clinical examinations did not reveal any symptoms of syndromic form of HL. The patient received cochlear implant at the age of 1 year and 5 months.
Auditory and speech performance in these cases were evaluated using the categories of auditory performance (CAP) [36] scale and speech intelligibility rating (SIR) [37] scale. These scales are reliable for measuring the outcome of cochlear implantation [38, 39]. CAP consists of 8 categories, ranging from “no awareness of environment” (CAP score 0) to “use of telephone with known users” (CAP score 7) and SIR consist of 5 categories, ranging from “unintelligible speech” (SIR score 1) to “speech intelligible to all listeners” (SIR score 5).
Molecular study
DNA extraction, GJB2 sequencing
Genomic DNA was extracted using Prime Prep Genomic DNA Extraction kit from blood (GeNet Bio, Korea), according to the manufacturer’s instruction. DNA purity and concentration was determined using the Nanodrop 2000 spectrophotometer (Nonodrop 2000 Thermo Scientific, USA) and its quality was checked on 1% agarose gel.
Sanger sequencing was performed in order to exclude GJB2 mutations and the following primers were used: F: 5′-CTCCCTGTTCTGTCCTAGCT-3′ and R: 5′-CTCATCCCTCTCATGCTGTC-3′ [40]. Because there was no positive family history of HL or other cognitive disorders in the family, linkage analysis was not sought.
Whole exome sequencing and bioinformatic analysis
The Sample was sent to Macrogen (South Korea) (https://www.macrogen.com/) and was subjected to WES using the Novaseq 4000 platform (Illumina, San Diego, CA, USA) with 151-bp paired-end reads. In summary, genomic DNA was fragmented to prepare Illumina library and fragments were captured to target all exons, splicing sites, and flanking intronic sequences of all genes (Agilent SureSelect V6 post). All fragments were amplified and then, sequencing was performed (the mean depth of coverage was 100X for greater than 92% of the sequences). For the studied sample, 57,006,242 reads were produced, and total read bases were 8.6G bp. The GC content was 52.01% and Q30 was 93.47%. After performing WES, the released raw data were converted to the FASTQ file. Bioinformatic analysis included GATK (Genome Analysis Toolkit) (https://gatk.broadinstitute.org/) for variant calling, BWA (Burrows-Wheeler Aligner) (http://bio-bwa.sourceforge.net/) for genome alignments and variant detection (hg19, NCBI Build 38) and Picard to mark duplicate reads were used. Variant filtering was performed based on Homozygous missense, start codon change, splice site, nonsense, stop loss, and indel variants with MAF < 1% in databases such as: dbSNP version 147, 1000 genomes project phase 3 database (https://www.internationalgenome.org/), NHLBI GO exome sequencing project (ESP) (https://evs.gs.washington.edu/), exome aggregation consortium (ExAC) (http://exac.broadinstitute.org) and Iranome (http://www.iranome.ir/).
After the filtration, the reported frameshift variant was evaluated by different in silico software tools such as PROVEAN (http://provean.jcvi.org/), PANTHER (http://www.pantherdb.org/), MutationTaster (http://www.mutationtaster.org/), SIFT (https://sift.bii.a-star.edu.sg/) and CADD (https://cadd.gs.washington.edu/) to predict its deleterious effect on protein in terms of function. Furthermore, the degree of conservation of this variants was assessed using NCBI BLAST of several vertebrate species [41].
Variant confirmation
The candidate variant was confirmed using bidirectional Sanger sequencing. Then, co-segregation analysis was performed using exon-specific custom primers to examine segregation of genotype and HL phenotype among the family members. PCR amplification and sequencing of this variant were performed using the forward primer: 5′-GCATCCATCTCCCATGTCTT-3′ and the reverse primer: 5′-CACCATCCAGCATCAGTC-3′ in exon 6. Chromatograms were compared with reference sequence (NM_001145308), encoding a 291 residue protein (NP_001138780.1), using SeqMan software version 5.00© (DNASTAR, Madison, WI, USA). Next, this variant was investigated in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/) and the literature to seek the novelty of the variant or its association with HL. Variant nomenclature was based on Human Genome Variation Society (HGVS) [41]. The American College of Medical Genetics and Genomics (ACMG) guidelines were also used to classify this variant [42]. The MEGA6 software was used to check the conservation of the mutated region in several species.
Results
Clinical evaluations
The proband was a four-year-old boy who showed bilateral profound NSHL, according to the audiological evaluations (Fig. 1c). Syndromic forms of HL were ruled out in this family, based on the history and clinical examination in the patient. The proband was born to a consanguineous first-cousin couple after a full-term natural delivery He showed no developmental delay or developmental regression, based on medical reports and examinations during pre-test genetic counseling. No genetic disease other than HL was evident in the related pedigree (Fig. 1). The CT scan results of Temporal Bone were normal in the patient.
Results of auditory and speech performance indicate good outcome of cochlear implantation in the patient after 3 years with CAP score of 6 (understanding conversation without lip reading) and SIR score of 5 (speech is intelligible to all listener).
Molecular findings
Direct sequencing of the coding exon of the GJB2 gene did not show any mutation. WES was applied and totally 672,262 variants were detected; one of them met the criteria for further analyses.
As a result of WES, a homozygous deletion of 16 nucleotides c.509_524del CAGTGGCTGAAAAACT (p.Ala170Alafs*20) in the LRTOMT gene was found. It causes frameshift in exon 6 of this gene. It creates alternation of 20 amino acids downstream of the deletion and leads to an early stop codon, resulting in a truncated protein with 170 residues (versus 291 residues in the intact protein). This deletion mutation was assessed as being deleterious by Mutation Taster as well as several other prediction tools such as SIFT, PROVEAN, PANTHER (Table 1). The frameshift variant was absent from dbSNP version 147, 1000 genomes project phase 3, NHLBI GO ESP, ExAC, Iranome, HGMD and Clinvar databases. It was not found in the literature, either. Ala170 and the following 20 amino acids that are modified in the mutated protein are located in a highly conserved residue of LRTOMT in multiple-species alignment (Fig. 3). It has been conserved among several species including Pan troglodytes, Macaca mulatta, Mus musculus, Rattus norvegicus and Xenopus tropicalis.
The variant co-segregated with the disease in the family: heterozygous in parents, but homozygous in the patient who was the only child of the family (Fig. 1d).
According to the ACMG guideline, that its evidence is describe below, this variant is classified as a pathogenic variant (1 very strong, 2 Moderate and 1 Supporting criteria):
-
It is a frameshift variant (a null variant) (PVS1).
-
It is located in a mutational hot spot and/or critical and well-established functional domain (PM1).
-
This variant is absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, Exome Aggregation Consortium and Iranome (local database) (PM2).
-
Multiple lines of computational evidence supported the deleterious effect of the variant on the gene or gene product (conservation, evolutionary, splicing impact, etc.) (PP3).
The 3-D structure of the LRTOMT protein (wild-type and mutated forms)
PDB files of the wild type and mutated protein were generated from protein structure prediction server (http://ps2.life.nctu.edu.tw/). Then, UCSF Chimera version 1.5.3 (https://www.cgl.ucsf.edu/chimera/) was used to construct the 3D structure of both wild-type and the mutant forms of LRTOMT. The pictures show a comparison between wild-type and mutant protein, the catalytic domain (catechol-O-methyltransferase) that is modified in the mutant protein (Fig. 2).

Protein structure modeling of wild-type and mutated LRTOMT. a the merged image of wild-type LRTOMT is shown in purple and mutanted LRTOMT in cyan. A part of the amino acid sequence has been eliminated in the mutated protein compared to the wild type protein (b) the catechol-O-methyltransferase domain, as a functional domain, is shown in yellow and in the figure (c) the modified COMT domain in the mutanted protein is defined in gray. The affected amino acids (residue 170–291) are a part of catalitic domain
Discussion
Hearing loss (HL) is a common sensorineural disorder, with an incidence of 1 in every 500–1000 children [43]. NSHL is a highly genetically heterogeneous neurosensory disorder with over 163 known genes (http://hereditaryhearingloss.org/) [44]. Although mutations in the GJB2 gene has been known to be the most common cause of ARNSHL in Iran (about 18.5% of HL cases), the role of other genes is remains to be illuminated [15, 45, 46]. Lack of accurate DNA diagnostics represents a real challenge for NSHL diagnosis and genetic counselling [47]. In this regard, NGS has addressed this problem with generation of huge data from our genes in a rather short time [48, 49].
In Iran, HL ranks second after intellectual disability [15]. To date, few studies have been performed on LRTOMT mutations in Iran and frequency of these mutations is not exactly known among Iranian ARNSHL patients [28, 50]. So far, about 20 pathogenic mutations have been reported in the LRTOMT gene (Table 2). The human LRTOMT gene (the DFNB63 locus) is located on the chromosome 11q13.2-q13.4 and encodes the LRTOMT protein. The locus DFNB63 was firstly mapped to the long arm of chromosome 11 by Kalay et al, who reported a five-generation Turkish family to be linked to this region [18]. Findings indicated that mouse Lrrc51 and Tomt are two separate genes encoding 2 different proteins, and that human LRTOMT gene is a larger fusion gene with two different transcripts [21]. The LRTOMT2 (residues 79 to 291) protein has a transmembrane catechol-O-methyltransferase (COMT) domain and is also known as COMT2, which is highly expressed in sensory hair cells and the vestibular organs of the inner ear [22]. The most important COMT domain is identified in the generic COMT protein that catalyzes the transfer of a methyl group getting from S-adenosyl methionine to catecholamines. It is associated with inactivation of catecholamine neurotransmitters like norepinephrine, dopamine and epinephrine [23, 57]. In a study in 2008, the mouse model of Comt2 mutation was generated and it was shown that mutation in this gene leads to profound HL and vestibular defect [22]. By using a zebrafish model, Erickson et al in 2017 showed that the defect in auditory and vestibular systems due to mutations in LRTOMT gene. It led to a lack of mechanotransduction (MET), a process in which sensory hair cells convert mechanical energy such as vestibular and auditory stimulation to electrical signals [58]. The TMC (Transmembrane channel-like) gene appears to be the most promising candidates to be the precursor of MET channel [59,60,61,62]. In humans, mutations in TMC1 gene are responsible for both recessive (DFNB7/11) and dominant (DFNA36) forms of NSHL [63]. Using the mercury mutant zebrafish, as a model of DFNB63, Erickson reported that LRTOMT is required for trafficking TMC proteins to the hair bundle [58, 64].
In several studies, it has been reported that NSHL due to mutations in the LRTOMT gene are more likely to be assigned to the LRTOMT2 (COMT2) region rather than LRTOMT1 [28, 51]. These findings indicate mutations in LRTOMT2 are associated with hair cell defects and lead to severe-to-profound NSHL [21, 29, 30, 53]. Patients with mutations in the LRTOMT gene have been reported exclusively from the Middle Eastern consanguineous families [19, 20, 28, 50]. The majority of reported mutations occurred in exon 5 and 7 of the LRTOMT2 coding region. Therefore, this region might be a mutational hot spot in the LRTOMT gene [9, 53].
In this study, a consanguineous family with a son suffering from bilateral sever-to-profound HL and negative for GJB2 gene mutations was selected for further study using WES. As a result, a novel long deletion variant (16 bp deletion) in the exon 6 of LRTOMT gene (NM_001145308) was found homozygously in the patient and heterozygously in the parents. It is a novel frameshift mutation (deletion of 16 nucleotides) in the LRTOMT gene, which has dual reading frames (ENST00000307198.7). In this transcript of LRTOMT gene, LRTOMT2 starts in exon 5 and ends in exon 7. The main portion of the catalytic domain (residues 79–291) of LRTOMT2 is eliminated as a result of this mutation (Fig. 2). The premature stop codon (20 codons after the position of the deletion) is predicted to result in a truncated protein with impaired function or no protein production, possibly because of nonsense mediated mRNA decay. The mutation region is a partly conserved in human and mice (Fig. 3), suggesting that the eliminated region is important for the catalytic functions of the enzyme.

The modified region (5 deleted amino acids shown in the red box and the folowing 20 residues that are changed due to the frameshift mutation) is located in a highly conserved region among species
Evaluation of auditory and speech performance in this study is in line with those of previous studies, suggesting that patients with LRTOMT mutations show good auditory performance after cochlear implant surgery [65].
Conclusions
In conclusion, our results suggest that mutations in the LRTOMT gene result in alterations in the LRTOMT2 (COMT2) protein and might be involved in sever-to-profound NSHL. The gene should be studied in a larger population of families in Iran for a more thorough understanding of its role in causing HL. This study showed that exome sequencing is an efficient molecular diagnostic method for ARNSHL as an extremely heterogeneous genetic disorder.
Availability of data and materials
The raw datasets generated and/or analyzed during the current study are not publicly available because it is possible that individual privacy could be compromised, but are available from the corresponding author on reasonable request.
Abbreviations
- HL:
-
Hearing loss
- ARNSHL:
-
Autosomal recessive non-syndromic hearing loss
- WES:
-
Whole exome sequencing
- ACMG:
-
American College of Medical Genetics and Genomics
- AR:
-
Autosomal recessive
- AD:
-
Autosomal dominant
- LRTOMT:
-
Leucine Rich Transmembrane and O-Methyl-Transferase
- COMT:
-
Catechol-O-methyltransferase
- OMIM:
-
Online Mendelian Inheritance in Man
- WGS:
-
Whole genome sequencing
- NGS:
-
Next generation sequencing
- TNGS:
-
Targeted next generation sequencing
- SNV:
-
Single nucleotide variants
- CNV:
-
Copy number variants
- CAP:
-
Categories of Auditory Performance
- SIR:
-
Speech Intelligibility Rating
- BWA:
-
Burrows-Wheeler Aligner
- GATK:
-
Genomic Analysis Toolkit
- MAF:
-
Minor Allele Frequency
- NHLBI:
-
National Heart, Lung, and Blood Institute
- ESP:
-
Exome Sequencing Project
- ExAC:
-
Exome Aggregation Consortium
- SIFT:
-
Scale-invariant feature transform
- CADD:
-
Combined Annotation Dependent Depletion
- NCBI:
-
National Center for Biotechnology Information
- BLAST:
-
Basic Local Alignment Search Tool
- PCR:
-
Polymerase chain reaction
- HGMD:
-
Human Gene Mutation Database
- HGVS:
-
Human Genome Variation Society
- PDB:
-
Protein data bank
- MET:
-
Mechanotransduction
References
Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354(20):2151–64.
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the US school-age population. Am J Med Genet. 1993;46(5):486–91.
Bouzid A, Smeti I, Dhouib L, Roche M, Achour I, Khalfallah A, et al. Down-expression of P2RX2, KCNQ5, ERBB3 and SOCS3 through DNA hypermethylation in elderly women with presbycusis. Biomarkers. 2018;23(4):347–56.
Bouzid A, Smeti I, Chakroun A, Loukil S, Gibriel AA, Grati M, et al. CDH23 Methylation status and presbycusis risk in elderly women. Front Aging Neurosci. 2018;10:241.
Atik T, Onay H, Aykut A, Bademci G, Kirazli T, Tekin M, et al. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS One. 2015;10(11):e0142154.
Petersen M, Willems P. Non-syndromic, autosomal-recessive deafness. Clin Genet. 2006;69(5):371–92.
Schrijver I. Hereditary non-syndromic sensorineural hearing loss: transforming silence to sound. J Mol Diagn. 2004;6(4):275–84.
Gorlin RJ, Gorlin RJ, Toriello HV, Cohen MM. Hereditary hearing loss and its syndromes. USA: Oxford University Press; 1995.
Charif M, Abidi O, Boulouiz R, Nahili H, Rouba H, Kandil M, et al. Molecular analysis of the TMPRSS3 gene in Moroccan families with non-syndromic hearing loss. Biochem Biophys Res Commun. 2012;419(4):643–7.
Stevens G, Flaxman S, Brunskill E, Mascarenhas M, Mathers CD, Finucane M. Global and regional hearing impairment prevalence: an analysis of 42 studies in 29 countries. Eur J Public Health. 2011;23(1):146–52.
Saadat M. Consanguineous marriages in Iranian folktales. Public Health Genom. 2007;10(1):38–40.
Rădulescu L, Mârţu C, Birkenhäger R, Cozma S, Ungureanu L, Laszig R. Prevalence of mutations located at the dfnb1 locus in a population of cochlear implanted children in eastern Romania. Int J Pediatr Otorhinolaryngol. 2012;76(1):90–4.
Najmabadi H, Cucci RA, Sahebjam S, Kouchakian N, Farhadi M, Kahrizi K, et al. GJB2 mutations in Iranians with autosomal recessive non-syndromic sensorineural hearing loss. Human Mutat. 2002;19(5):572.
Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Human Genet. 2005;77(6):945–57.
Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Human Genet. 2010;55(10):639.
Bazazzadegan N, Nikzat N, Fattahi Z, Nishimura C, Meyer N, Sahraian S, et al. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss—a twelve year study. Int J Pediatr Otorhinolaryngol. 2012;76(8):1164–74.
Sirmaci A, Edwards YJ, Akay H, Tekin M. Challenges in whole exome sequencing: an example from hereditary deafness. PLoS One. 2012;7(2):e32000.
Kalay E, Caylan R, Kıroglu A, Yasar T, Collin R, Heister J, et al. A novel locus for autosomal recessive nonsyndromic hearing impairment, DFNB63, maps to chromosome 11q13. 2–q13. 4. J Mol Med. 2007;85(4):397–404.
Khan SY, Riazuddin S, Tariq M, Anwar S, Shabbir MI, Riazuddin SA, et al. Autosomal recessive nonsyndromic deafness locus DFNB63 at chromosome 11q13. 2–q13. 3. Hum Genet. 2007;120(6):789–93.
Tlili A, Masmoudi S, Dhouib H, Bouaziz S, Rebeh IB, Chouchen J, et al. Localization of a Novel Autosomal Recessive Non-syndromic Hearing Impairment Locus DFNB63 to Chromosome 11q13. 3-q13. 4. Ann Hum Genet. 2007;71(2):271–5.
Ahmed ZM, Masmoudi S, Kalay E, Belyantseva IA, Mosrati MA, Collin RW, et al. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet. 2008;40(11):1335.
Du X, Schwander M, Moresco EMY, Viviani P, Haller C, Hildebrand MS, et al. A catechol-O-methyltransferase that is essential for auditory function in mice and humans. Proc Natl Acad Sci. 2008;105(38):14609–14.
Bonifácio MJ, Archer M, Rodrigues ML, Matias PM, Learmonth DA, Carrondo MA, et al. Kinetics and crystal structure of catechol-o-methyltransferase complex with co-substrate and a novel inhibitor with potential therapeutic application. Mol Pharmacol. 2002;62(4):795–805.
Lautala P, Ulmanen I, Taskinen J. Molecular mechanisms controlling the rate and specificity of catechol O-methylation by human soluble catechol O-methyltransferase. Mol Pharmacol. 2001;59(2):393–402.
Zhu B. Catechol-O-Methyltransferase (COMT)-mediated methylation metabolism of endogenous bioactive catechols and modulation by endobiotics and xenobiotics: importance in pathophysiology and pathogenesis. Curr Drug Metab. 2002;3(3):321–49.
Gregory-Evans CY, Moosajee M, Hodges MD, Mackay DS, Game L, Vargesson N, et al. SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Hum Mol Genet. 2007;16(20):2482–93.
Tekin M, Hişmi BÖ, Fitoz S, Özdağ H, Cengiz FB, Sırmacı A, et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007;80(2):338–44.
Babanejad M, Fattahi Z, Bazazzadegan N, Nishimura C, Meyer N, Nikzat N, et al. A comprehensive study to determine heterogeneity of autosomal recessive nonsyndromic hearing loss in Iran. Am J Med Genet Part A. 2012;158(10):2485–92.
Gibriel AA, Abou-Elew MH, Masmoudi S. Analysis of p. Gly12Valfs* 2, p. Trp24* and p. Trp77Arg mutations in GJB2 and p. Arg81Gln variant in LRTOMT among non syndromic hearing loss Egyptian patients: implications for genetic diagnosis. Mol Biol Rep. 2019;46(2):2139–45.
Chakchouk I, Said MB, Jbeli F, Benmarzoug R, Loukil S, Smeti I, et al. NADf chip, a two-color microarray for simultaneous screening of multigene mutations associated with hearing impairment in North African Mediterranean countries. J Mol Diagn. 2015;17(2):155–61.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333.
Schrauwen I, Helfmann S, Inagaki A, Predoehl F, Tabatabaiefar MA, Picher MM, et al. A mutation in CABP2, expressed in cochlear hair cells, causes autosomal-recessive hearing impairment. Am J Hum Genet. 2012;91(4):636–45.
Woo H-M, Park H-J, Baek J-I, Park M-H, Kim U-K, Sagong B, et al. Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet. 2013;14(1):72.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368.
Wu CC, Liu TC, Wang SH, Hsu CJ, Wu CM. Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope. 2011;121(6):1287–93.
Fang H-Y, Ko H-C, Wang N-M, Fang T-J, Chao W-C, Tsou Y-T, et al. Auditory performance and speech intelligibility of Mandarin-speaking children implanted before age 5. Int J Pediatr Otorhinolaryngol. 2014;78(5):799–803.
Archbold S, Lutman ME, Nikolopoulos T. Categories of auditory performance: inter-user reliability. Br J Audiol. 1998;32(1):7–12.
Allen C, Nikolopoulos TP, Dyar D, O’Donoghue GM. Reliability of a rating scale for measuring speech intelligibility after pediatric cochlear implantation. Otol Neurotol. 2001;22(5):631–3.
Tabatabaiefar MA, Alasti F, Zohour MM, Shariati L, Farrokhi E, Farhud D, et al. Genetic linkage analysis of 15 DFNB loci in a group of Iranian families with autosomal recessive hearing loss. Iranian J Public Health. 2011;40(2):34.
den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405.
Yazdanpanahi N, Tabatabaiefar MA, Farrokhi E, Abdian N, Bagheri N, Shahbazi S, et al. Compound heterozygosity for two novel SLC26A4 mutations in a large Iranian pedigree with Pendred syndrome. Clin Exp Otorhinolaryngol. 2013;6(4):201.
Van Camp G, Smith RJ. Hereditary hearing loss homepage; 2006.
Hashemzadeh Chaleshtori M, Farhud D, Patton M. Familial and sporadic GJB2-related deafness in Iran: review of gene mutations. Iran J Public Health. 2007;36(1):1–14.
Mahdieh N, Rabbani B, Shirkavand A, Bagherian H, Movahed ZS, Fouladi P, et al. Impact of consanguineous marriages in GJB2-related hearing loss in the Iranian population: a report of a novel variant. Genet Test Mol Biomarkers. 2011;15(7–8):489–93.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–11.
Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461(7261):272.
Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3(87):87re3.
Taghizadeh SH, Kazeminezhad SR, Sefidgar SAA, Yazdanpanahi N, Tabatabaeifar MA, Yousefi A, et al. Investigation of LRTOMT gene (locus DFNB63) mutations in Iranian patients with autosomal recessive non-syndromic hearing loss. Int J Mol Cell Med. 2013;2(1):41.
Vanwesemael M, Schrauwen I, Ceuppens R, Alasti F, Jorssen E, Farrokhi E, et al. A 1 bp deletion in the dual reading frame deafness gene LRTOMT causes a frameshift from the first into the second reading frame. Am J Med Genet Part A. 2011;155(8):2021–3.
Wang R, Han S, Khan A, Zhang X. Molecular analysis of twelve Pakistani families with nonsyndromic or syndromic hearing loss. Genet Test Mol Biomarkers. 2017;21(5):316–21.
Ichinose A, Moteki H, Hattori M, Nishio SY, Usami SI. Novel mutations in LRTOMT associated with moderate progressive hearing loss in autosomal recessive inheritance. Ann Otol Rhinol Laryngol. 2015;124(1_suppl):142S–7S.
Riahi Z, Bonnet C, Zainine R, Louha M, Bouyacoub Y, Laroussi N, et al. Whole exome sequencing identifies new causative mutations in Tunisian families with non-syndromic deafness. PLoS One. 2014;9(6):e99797.
Marková S, Brožková DŠ, Mészárosová A, Neupauerová J, Groh D, Křečková G, et al. Mutations in eight small DFNB genes are not a frequent cause of non-syndromic hereditary hearing loss in Czech patients. Int J Pediatr Otorhinolaryngol. 2016;86:27–33.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50.
Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60(2):141–51.
Erickson T, Morgan CP, Olt J, Hardy K, Busch-Nentwich E, Maeda R, et al. Integration of Tmc1/2 into the mechanotransduction complex in zebrafish hair cells is regulated by Transmembrane O-methyltransferase (Tomt). Elife. 2017;6:e28474.
Corey DP, Holt JR. Are TMCs the mechanotransduction channels of vertebrate hair cells? J Neurosci. 2016;36(43):10921–6.
Wu Z, Müller U. Molecular identity of the mechanotransduction channel in hair cells: not quiet there yet. J Neurosci. 2016;36(43):10927–34.
Beurg M, Kim KX, Fettiplace R. Conductance and block of hair-cell mechanotransducer channels in transmembrane channel–like protein mutants. J Gen Physiol. 2014;144(1):55–69.
Pan B, Géléoc GS, Asai Y, Horwitz GC, Kurima K, Ishikawa K, et al. TMC1 and TMC2 are components of the mechanotransduction channel in hair cells of the mammalian inner ear. Neuron. 2013;79(3):504–15.
Kurima K, Peters L, Yang Y, Riazuddin S, Ahmed Z, Naz S, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30:277–84.
Kawashima Y, Géléoc GS, Kurima K, Labay V, Lelli A, Asai Y, et al. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel–like genes. J Clin Invest. 2011;121(12):4796–809.
Wu C-C, Lin Y-H, Liu T-C, Lin K-N, Yang W-S, Hsu C-J, et al. Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine. 2015;94(27):e1073.
Acknowledgments
We take this opportunity to express our special thanks to the staff at the Isfahan Cochlear Implantation Center and also to the patient and family.
Funding
This work was financially supported by Isfahan University of Medical Sciences. Whole exome sequencing and its data collection and data analysis were supported by Isfahan University of Medical Sciences (grant NO.396133 and NO.394805). The funding bodies played no role in in study design and writing the manuscript.
Author information
Authors and Affiliations
Contributions
Study design: MAT; Enrolled the patient and contributed to clinical diagnoses and report writing: MS, MHN, HA; Data analysis and interpretation: MAT, AS, SN1, MSF, SN2, ZN; Manuscript preparation: AS, SN1, ZN; critically reviewed by MAT. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Review Board of Isfahan University of Medical Sciences (grant no: 396133 and 394805). Written informed consent was obtained from all of the participants in the study and a written consent to participate was obtained from the parents of the patient (younger than the age of 16).
Consent for publication
Written informed consent for publication of clinical details and clinical images was obtained from the all of the participants and from the parents the participant under the age of 18.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sarmadi, A., Nasrniya, S., Soleimani Farsani, M. et al. A novel pathogenic variant in the LRTOMT gene causes autosomal recessive non-syndromic hearing loss in an Iranian family. BMC Med Genet 21, 127 (2020). https://doi.org/10.1186/s12881-020-01061-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01061-7