
Abstract
Background
Spinal muscular atrophy (SMA) is a rare neuromuscular disorder threating hundreds of thousands of lives worldwide. And the severity of SMA differs among different clinical types, which has been demonstrated to be modified by factors like SMN2, SERF1, NAIP, GTF2H2 and PLS3. However, the severities of many SMA cases, especially the cases within a family, often failed to be explained by these modifiers. Therefore, other modifiers are still waiting to be explored.
Case presentation
In this study, we presented a rare case of SMA discordant family with a mild SMA male patient and a severe SMA female patient. The two SMA cases fulfilled the diagnostic criteria defined by the International SMA Consortium. With whole exome sequencing, we confirmed the heterozygous deletion of exon7 at SMN1 on the parents’ genomes and the homozygous deletions on the two patients’ genomes. The MLPA results confirmed the deletions and indicated that all the family members carry two copies of SMN2, SERF1, NAIP and GTF2H2. Further genomic analysis identified compound heterozygous mutations at TLL2 on the male patient’s genome, and compound heterozygous mutations at VPS13A and the de novo mutation at AGAP5 on female patient’s genome. TLL2 is an activator of myostatin, which negatively regulates the growth of skeletal muscle tissue. Mutation in TLL2 has been proved to increase muscular function in mice model. VPS13A encodes proteins that control the cycling of proteins through the trans-Golgi network to endosomes, lysosomes and the plasma membrane. And AGAP5 was reported to have GTPase activator activity.
Conclusions
We reported a case of SMA discordant family and identified mutations at TLL2, VPS13A and AGAP5 on the patients’ genomes. The mutations at TLL2 were predicted to be pathogenic and are likely to alleviate the severity of the male SMA patient. Our finding broadens the spectrum of genetic modifiers of SMA and will contribute to accurate counseling of SMA affected patients and families.
Similar content being viewed by others


Background
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease characterized by degeneration of motor neurons of the spinal, which affects 1 in 6000 to 1 in 10,000 individuals worldwide [1]. Based on the age of onset and the highest motor function the patient could achieve, SMA has been divided into four clinical types: severe type I (Werdnig-Hoffmann disease, OMIM:253300), intermediate type II (OMIM:253550), mild type III (Kugelberg-Welander syndrome, OMIM:253400), and adult-onset type IV (OMIM:271150) [2]. It has been reported that about 60% of newborn SMA patients belong to the severe type I SMA [3]. Homozygous mutations of the survival motor neuron 1 gene (SMN1) is the main cause of all types of SMA (accounting for over 95% of cases). It has been reported that the severity of SMA is mainly modified by SMN2 gene copy number. About 80% of patients with type I SMA have one or two SMN2 copies, 82% of type II SMA patients have two or three SMN2 copies, 96% of patients with type III SMA have three or four SMN2 copies and 75% of type IV SMA patients harbor four SMN2 copies [4, 5]. Besides, SERF1(H4F5), NAIP and GTF2H2(p44) locating in close to SMN locus have also been related to SMA severity [6,7,8]. However, the severities of many SMA cases, especially the cases within a family, often failed to be explained by these modifiers, indicating the existence of other genes modifying the symptoms of SMA [9]. Recently, increasing evidence shows that additional factors, such as proteins interacting with SMN, DNA methylation level, factors influencing SMN2 expression, may contribute to SMA phenotype modification. Among them, the most well-known factors are Plastin 3 (PLS3), zinc finger protein 1 (ZPR1) and Neuritin 1 (NRN1). The expression levels of these factors were found decreased in SMA patients and overexpression of them could rescue the SMA phenotypes [10,11,12,13]. In this study, we present a SMA discordant family with a mild SMA male patient and a severe female patient, and analyzed them with whole exome sequencing. We try to identify genomic factors that confer the phenotype discordance within the family, which will help us understand the pathophysiology of SMA and discover potential therapeutic targets for neuromuscular diseases.
Case presentation
Here, we present a case of SMA-discordant family with healthy parents and a SMA affected male-female sib pair. The sibs have different clinical manifestations and the female patient showed significant severe phenotypes (Table 1). The male patient was a 16-year-old boy, the first child of East Asian healthy non-consanguineous parents. He was born after an uneventful pregnancy by normal spontaneous delivery at the 39th week of gestation. His birth weight was 3400 g (75th centile) and length was 50 cm (50–75th centile). He was healthy at birth and could sit and walk unaided after 1 year old. But he showed mild SMA symptoms at the age of 3. During infancy, his medical history was unremarkable. The female patient was 11-year-old girl. She was born by normal spontaneous delivery at the 37th week of gestation. Her birth weight was 2900 g (50th centile) and length was 50 cm (50–75th centile). In contrast to her brother, the girl was floppy at birth and needed respiratory support duo to dyspnea. She was never able to sit and stand unaided, and died due to pneumonia at the age of 11. During infancy and childhood, the female patient was admitted to hospital several times duo to respiratory infections. The intelligence of the two patients were normal but they did not attend school because of their disabilities. The detailed symptoms of the patients are shown in Table 1.
Discussion and conclusions
In this study, we present a rare case of atypical SMA discordant family with a male patient diagnosed with mild type III SMA and a female patient diagnosed with severe type I SMA. To identify the genomic difference between two SMA patients and find the genetic basis conferring phenotype differences, we sequenced the two patients and their parents with whole exome sequencing. The sequencing reads were mapped to the reference genome of hg19 with bwa [14] and the alignments showed the heterozygous deletion of exon7 at SMN1 on the parents’ genomes and the homozygous deletion on the two patients’ genomes, confirming that the SMA of the two patients were caused by SMN1 mutation (Fig. 1 and Additional file 1: Figure S1). Previous studies showed that about 82% of type II SMA patients have two or three SMN2 copies. In our study, all the family members have two copies of SMN2, and the male patient is affected by mild SMA and the female patient was affected by severe SMA. Besides SMN2, SERF1, NAIP and GTF2H2 also have been reported to affect the symptoms of SMA. However, in our case, no sequence difference (Fig. 2) and copy number variations (Additional file 1: Figure S1) were identified in the three modifiers between the two patients. Therefore, there could be other modifiers that contribute to the phenotype discordance. To find the genomic differences that contribute to the phenotype differences, we inferred the high-quality variants in the four samples and analyzed them in three possible inheritance modes, including autosomal recessive model, de novo model and compound heterozygous model (Fig. 3). We identified compound heterozygous mutations at TLL2 on the male patient’s genome, and compound heterozygous mutations at VPS13A and the de novo mutation at AGAP5 on female patient’s genome (Additional file 1: Figures S2-S6). All the five variants were also confirmed by Sanger sequencing (Additional file 1: Figure S7) with the primer showed in Additional file 1: Table S1. The variants on the girl’s genome were not verified because of her death.

Reads coverages in SMN exon regions. The four family members were sequenced by WES and the reads were aligned to the exon regions of SMN1 (a) and SMN2 (b). The grey peaks indicate the richness of the reads that mapped to the corresponding exon region in each sample. The red box indicates the alignment coverages of exon 7 of SMN1 and SMN2 in each sample

Genetic map of SMA related locus and reads coverages of candidate SMA modifiers. a SMN and their surrounding genes are contained within two large inverted genomic fragments within the region on chromosome 5q13. SMN1 is located within the telomeric copy whereas SMN2 is contained within the centromeric copy. The surrounding genes include SERF1, NAIP and GTF2H2, which are reported as candidate modifiers in SMA. The arrows indicate their directions. b, c, d The reads coverages in the b SERF1, c NAIP and d GTF2H2 exon regions for each family member. The grey peaks indicate the richness of the reads that mapped to the corresponding exon region in each sample

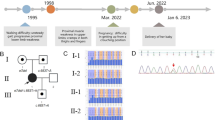
Pedigree and WES analysis strategy of SMA-discordant family. a The pedigree of the family. The man (layer II) married a woman (layer II) who came from the same village and they had two SMA affected children. The male (layer III) was diagnosed with type III SMA (gray) and the female was diagnosed with type I SMA (black). The female patient died at the age of 11. The parents and the two patients were sequenced by WES. And compound heterozygous mutations in TLL2 were identified on the male patient’s genome. b Variants filtering pipeline of the analysis. Sentieon (https://www.sentieon.com) and Strelka2 (https://github.com/Illumina/strelka) were used to call the variants of the four samples. And the variants that called by both tools were kept for the further analysis. The variants were analyzed with GEMINI (https://github.com/arq5x/gemini). N/M represents that N variants were identified on the male patient’s genome and M variants were identified on the female patient’s genome
The effects of variants identified in TLL2, AGAP5 and VPS13A were predicted with 6 tools (Additional file 1: Table S2) including SIFT [15], Polyphen2 [16], CADD [17], M-CAP [18], DANN [19] and FATHMM-MKL [20]. All the variants except c.1393A > G of AGAP5 were predicted to be damaging by more than two tools. Furthermore, except variant of AGAP5, the CADD scores of all the other variants are more than 10, which means they are among the top 1% variants and are likely to have deleterious effects. The variant c.1609C > T of TLL2 was predicted as a damage mutation by all the 6 tools except M-CAP. And the variant c.112G > C of TLL2 was predicted to be pathogenic by M-CAP, DANN and FATHMM-MKL. The c.1393A > G of AGAP5 was predicted to be pathogenic by Polyphen2. And the c.4174A > T of VPS13A was predicted be pathogenic by Polyphen2, DANN and FATHMM-MKL. The c.5728A > G of VPS13A was predicted be pathogenic by DANN and FATHMM-MKL. According to ACMG 2015 guidelines [21], the variants of TLL2 and AGAP5 were classified as likely pathogenic variants and the other two variants were classified as variants of uncertain significance (Additional file 1: Table S2). The variants of c.112G > C and c.1609C > T in TLL2 have been submitted to ClinVar under the accession SCV000920794 and SCV000920793. All the rare variants (AF < 0.05) detected in this study were listed in Additional file 2: Table S3.
SMN2, SERF1, NAIP and GTF2H2 have been reported to affect the severities of SMA. However, in our SMA discordant family, no sequence difference and copy number variations were found in these modifiers between the two patients. Therefore, we hypothesize that there may exist other genomic factors that confer the phenotype differences between the two patients. By WES and sequence analysis, we identified 2 pathogenic variants in TLL2 on the male patient’s genome and 3 high impact variants involving in AGAP5 and VPS13A on the female patient’s genome. TLL2 encodes a proteinase in the BMP-1/TLD protein family and is capable of activating myostatin. Lee showed that the knockout of TLL2 in mice increased the muscle mass and improved SMA [22]. The target of TLL2, myostatin, is a myokine that inhibits muscle cell growth and differentiation. And it has been proposed to be a drug target for SMA and the TOPAZ, an inhibitor of the activation of myostatin, has entered into phase 2 clinical trial (NCT03921528). Recently Long et al. showed that the inhibition of myostatin was beneficial in SMA mice models [23]. In addition to TLL2, VPS13A and AGAP5 were found to have mutations on the female’s genome. VPS13A encodes proteins that control the cycling of proteins through the trans-Golgi network to endosomes, lysosomes and the plasma membrane. It has been reported to involve in Choreoacanthocytosis (OMIM:200150), however, the age of onset for Choreoacanthocytosis is 26 to 59 years old [24], suggesting that the female was less likely to be affected by VPS13A mutations. AGAP5 encodes a protein annotated with GTPase activator activity and its function in SMA is unknown.
It has been reported that the expression level of PLS3, ZPR1 or NRN1 is correlated with the SMA severity. Yener et al. stated that the expression levels of PLS3 and NRN1 were different between the patients from SMA discordance families. However, their findings also show that PLS3 and NRN1 expressions do not always modify SMA phenotype [10]. In our study, the phenotype discordance could be modified by PLS3, ZPR1 or NRN1 on expression level. But, the genomic difference between the two patients from SMA discordance will provide us a new insight into understanding of SMA pathophysiology.
Because of technology limitation of the WES, the phenotype difference of the two patients in our study may be caused by the undetected mutations in the reported modifiers. In addition, other mutations, such as non-coding mutation and structural variation, could also contribute to the phenotype difference. Therefore, our study in some extend should be considered as preliminary research and the function of the variants should be confirmed by functional studies. The expression level of factors like PLS3, ZPR1 and NRN1 were not detected in our study because of the female patient’s death. We hope our study could provide a new insight into SMA pathophysiology in genomic view.
In conclusion, we have presented a case of SMA discordant family with an affected male-female sib pair and identified compound heterozygous mutations at TLL2 on the male patient’s genome, and compound heterozygous mutations at VPS13A and the de novo mutation at AGAP5 on female patient’s genome. The pathogenic mutations at TLL2 are likely to affect the severity of the SMA. Our findings add new knowledge to SMA and will contribute to accurate counseling of SMA affected patients and families.
Availability of data and materials
The sequence datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised. However, datasets are available from the corresponding author on reasonable request.
Abbreviations
- AF:
-
Allele frequency
- MLPA:
-
Multiplex ligation-dependent probe amplification
- OMIM:
-
Online mendelian inheritance in man
- SMA:
-
Spinal muscular atrophy
- WES:
-
Whole exome sequencing
References
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–33.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online Mendelian inheritance in man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43(Database issue):D789–98.
Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, Cook SF, Lochmuller H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124.
Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–68.
Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, Matsuo M, Nishio H. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol. 2002;249(9):1211–9.
Scharf JM, Endrizzi MG, Wetter A, Huang S, Thompson TG, Zerres K, Dietrich WF, Wirth B, Kunkel LM. Identification of a candidate modifying gene for spinal muscular atrophy by comparative genomics. Nat Genet. 1998;20(1):83–6.
Roy N, Mahadevan MS, McLean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner-Johnston A, Lefebvre C, Kang X, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80(1):167–78.
Burglen L, Seroz T, Miniou P, Lefebvre S, Burlet P, Munnich A, Pequignot EV, Egly JM, Melki J. The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am J Hum Genet. 1997;60(1):72–9.
Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831–46.
Yener IH, Topaloglu H, Erdem-Ozdamar S, Dayangac-Erden D. Transcript levels of plastin 3 and neuritin 1 modifier genes in spinal muscular atrophy siblings. Pediatr Int. 2017;59(1):53–6.
Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet. 2012;21(12):2745–58.
Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–7.
Akten B, Kye MJ, le Hao T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, et al. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A. 2011;108(25):10337–42.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581–6.
Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31(5):761–3.
Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–3.
Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med. 2016;18(11):1165.
Lee SJ. Genetic analysis of the role of proteolysis in the activation of latent myostatin. PLoS One. 2008;3(2):e1628.
Long KK, O'Shea KM, Khairallah RJ, Howell K, Paushkin S, Chen KS, Cote SM, Webster MT, Stains JP, Treece E, et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet. 2019;28(7):1076–89.
Rubio JP, Danek A, Stone C, Chalmers R, Wood N, Verellen C, Ferrer X, Malandrini A, Fabrizi GM, Manfredi M, et al. Chorea-acanthocytosis: genetic linkage to chromosome 9q21. Am J Hum Genet. 1997;61(4):899–908.
Acknowledgements
The authors would like to thank Dr. Peining Li and Dr. Gang Peng in the department of genetics at Yale university for valuable discussion.
Funding
During the whole course of this study, the study design, data analysis, data interpretation and manuscript preparation were mainly supported by National Key R&D Program of China(Grand No. 2018YFC0910500), the Neil Shen’s SJTU Medical Research Fund, SJTU-Yale Collaborative Research Seed Fund, National Natural Science Foundation of China (Grand No. 31728012) and Science and Technology Commission of Shanghai Municipality (Grant No. 17DZ22512000). Data collection and part of the data interpretation were supported by Lishui medical key disciplines (Grand No. 2016zdxk06) and Zhejiang laboratory animal science and technology program (Grand No. 2016C37145).
Author information
Authors and Affiliations
Contributions
JJ, JG and XC analyzed the sequencing data. JJ interpreted the data and wrote the manuscript. JH analyzed the patients’ medical information data, collected the blood specimens and interpreted the data. HZ and HL conceived and designed the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all participants or their parents (for minors), and the Ethics Committee of the Sixth Affiliated Hospital of Wenzhou Medical University approved this study.
Consent for publication
Written informed consent was obtained from all participants or their parents (for minors) included in this report for the use of clinical-related materials for scientific research and publications. These materials include medical information, genetic testing results and other related data used in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Supplmentary materials for this study.
Additional file 2:
Table S3. The list of variants that have a minor allele frequency less than 5% in 1000 Genomes or ESP-6500.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, J., Huang, J., Gu, J. et al. Genomic analysis of a spinal muscular atrophy (SMA) discordant family identifies a novel mutation in TLL2, an activator of growth differentiation factor 8 (myostatin): a case report. BMC Med Genet 20, 204 (2019). https://doi.org/10.1186/s12881-019-0935-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0935-3