
Abstract
Background
Ovarian cancer (OC) is the most deadly gynaecological cancer, contributing significantly to female cancer-related deaths worldwide. Improving the outlook for OC patients depends on the identification of more reliable prognostic biomarkers for early diagnosis and survival prediction. The various roles of long non-coding RNAs (lncRNAs) in OC have attracted increasing attention. This study aimed to identify a lncRNA-based signature for survival prediction in OC patients.
Methods
RNA expression data and clinical information from a large number of OC patients were downloaded from a public database. These data were regarded as a training set to construct a weighed gene co-expression network analysis (WGCNA) network, mine stable modules, and screen differentially expressed lncRNAs. The prognostic lncRNAs were screened using univariate Cox regression analysis and the optimal prognosis lncRNA combination was screened using a Cox-PH model. The finalised lncRNA combination was used to construct the risk score system, which was validated and assessed for effectiveness using other independent datasets. Further functional pathway enrichment was performed using gene set enrichment analysis (GSEA).
Results
A co-expression network was constructed and four stable modules with OC-related biological functions were obtained. A total of 19 lncRNAs significantly related to prognosis of ovarian cancer were obtained using univariate Cox regression analysis, and the 5 prognostic signature lncRNAs GAS5, HCP5, PART1, SNHG11, and SNHG5 were used to establish a risk assessment system. The reliability of the prognostic scoring system was further confirmed using validation sets, which indicated that the risk assessment system could be used as an independent prognostic factor. Pathway enrichment analysis revealed that the network modules related to the above five prognostic genes were significantly associated with cell local adhesion, cancer signaling pathways, JAK-STAT signalling, and endogenous cell receptor interaction.
Conclusions
The risk score system established in this study could provide a novel reliable method to identify individuals at high risk of OC. In addition, the five prognostic lncRNAs identified here are promising potential prognostic biomarkers that could help to elucidate the pathogenesis of OC.
Similar content being viewed by others

Background
Ovarian cancer (OC) is the most deadly gynaecological cancer and a primary cause of female cancer-related deaths worldwide, with recurrent OC being incurable in almost all cases [1, 2]. Due to the mild or absent signs and symptoms during early stages and the lack of reliable early detection tests, OC is usually diagnosed at the late stages, leading to poor prognosis and a five-year survival rate of only 30% [3, 4]. Therefore, a deeper understanding of its regulatory machinery at the molecular level is critical to identify reliable prognostic biomarkers for early diagnosis and survival prediction in OC patients.
Long non-coding RNAs (lncRNAs) are a group of non-coding RNAs longer than 200 nucleotides. Growing bodies of evidence have demonstrated the involvement of lncRNAs in OC, with at least 56 OC-related lncRNAs having been identified so far [4]. LncRNAs exhibit multiple biological functions during the various stages of OC development, and their deregulated expression is closely associated with OC early diagnosis, prognosis and response to chemotherapy [4,5,6,7,8,9]. Some lncRNAs, such as NEAT1 and GAS5, have been identified as clinical prognostic biomarkers, and are being used as potential therapeutic targets for OC [10,11,12]. Although previous studies have made remarkable progress, the prognostic roles of lncRNAs in OC and the related underlying mechanisms remain poorly characterized. Further research efforts are needed to identify, characterize, and elucidate the detailed functions of lncRNAs at the molecular level, and to identify more lncRNAs related to the prognosis of OC.
Recently, models for disease risk prediction and subsequent prognostic evaluation have attracted increasing interest from researchers, as it has become clear that the pathogenesis of most diseases is mediated by multiple, rather than single genes [13,14,15,16]. Risk assessment tools can help to estimate the probability that an individual with a given set of risk factors will develop a disease of interest, as well as detect high-risk populations for a given disease [15, 16]. So far, such risk assessment tools have been widely applied to the clinical prediction of various cancers [15, 17, 18], resulting in the identification of several expression-based lncRNA signatures as risk assessment tools for OC [3, 6, 9, 19]. However, as the risk assessment tools for OC are still limited, more research is necessary to more exhaustively establish a set of reliable tools for risk prediction.
In the present study, RNA expression data and clinical information from a large number of OC patients were downloaded from a public database, and a co-expression network was built to excavate network modules with OC-related biological functions. LncRNAs in stable functional modules were identified as important factors associated with OC. Combined with the clinical survival and prognostic information of OC samples; we identified a molecular lncRNA combination, which was significantly associated with OC prognosis. Based on these prognostic-related lncRNAs, a prognostic risk assessment model was constructed to identify significant differences between high-risk and low-risk prognostic samples. The reliability of the model was validated using the clinical survival and prognostic information of OC samples in independent validation datasets.
Methods
Data sources
The mRNA-seq expression profiles of OC in the training dataset were downloaded from the TCGA database (https://portal.gdc.cancer.gov/). A total of 419 ovarian cancer samples were detected by the Illumina HiSeq 2000 RNA sequencing platform. For the validation datasets, we first used the key word “ovarian cancer” to search all publicly uploaded expression data in the NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) database. The following criteria were then used for inclusion into the single validation datasets: 1) all data contained gene expression profiles; 2) detection objects were solid tumour tissues from patients with OC, exclusive of blood and cell lines; 3) expression profiles were all from human subjects; and 4) the sample number was no less than 40. The two final datasets GSE32062 and GSE17260 satisfied all the requirements, and included 260 and 100 samples, respectively.
Data pre-processing
The downloaded data were pre-processed before further analysis. For the data downloaded from the TCGA database, normalisation was performed using the quantile standardization method of pre-process Core version 1.40.0 package [20] in R3.4.1 language (http://bioconductor.org/packages/release/bioc/html/preprocessCore.html). Next, lncRNAs were annotated using Ref_seq and Transcript_ID of the annotation platform and aligned by Blast to human genome sequences (GRCh38 version) using Clustal2 [21] (http://www.clustal.org/clustal2/). Finally, lncRNAs and their corresponding expression values were calculated [19].
For the expression profiles with CEL original format, data were converted into expression values using the oligoversion 1.41.1 package [22] in R3.4.1 language (http://www.bioconductor.org/packages/release/bioc/html/oligo.html). Missing data were supplemented with median values, followed by background correction using MAS methods and normalisation using quantile methods. For the expression profiles with. TXT original format, data were log2 transformed and normalised using the median standardization method in limma version 3.34.0 [23] in R3.4.1 language (https://bioconductor.org/packages/release/bioc/html/limma.html).
Selection of stable modules using WGCNA
Weighed gene co-expression network analysis (WGCNA), a bioinformatics algorithm for construction of co-expression networks, is commonly used to identify modules associated with diseases and consequently screen important pathogenic mechanisms or potential therapeutic targets [24]. A co-expression network was constructed using the WGCNA version 1.61 package [25] in R3.4.1 language (https://cran.r-project.org/web/packages/WGCNA/index.html) based on the TCGA datasets.
WCGNA analysis needed to satisfy the prerequisite of scale-independent network distribution. Hence, the appropriate weight parameter β (power) of the adjacency matrix needed to be selected to ensure the constructed co-expression network approached the scale-free distribution to the greatest extent. With 1~20 of β value ranges, the linear model was established by logarithms of the adjacency degree of a node (log k) and the appearance probability of this node [log(p(k))]. The β parameters were the square values of R coefficients. A higher R2 value indicated that the network was closer to the scale-independent distribution. The β (power) value when R2 was approximately 0.9 for the first time was finally chosen for further analysis. All genes were ranked according to their expression values using a rank function. Then, the correlation between each pair of datasets of the three expression profiles (TCGA, GSE32062 and GSE17260) was evaluated using the verboseScatterplot function and RNA adjacency matrix to construct an RNA correlation matrix. This matrix was used as the basis to build the hierarchical clustering tree using the criteria of a cut height of 0.99, and the involvement of at least 200 RNAs per module. Finally, the userListEnrichment function was used to identify stable modules associated with OC using the criteria of a Z-score greater than 5. The lncRNAs in the stable modules were defined as those significantly associated with OC.
Construction of the risk prediction model
The lncRNAs associated with prognosis were further identified by univariate Cox regression analysis of the survival package in R3.4.1 language. A P value less than 0.05 obtained by the log-rank test was chosen as the threshold for identification. Next, the optimal combinations of prognostic-related lncRNAs were screened using the Cox-Proportional Hazards (Cox-PH) model [26] based on L1-penalised regularization regression algorithm of the penalised package [27] in R3.4.1 language (http://bioconductor.org/packages/penalised/). The optimized parameter “lambda” in the screening model was obtained by 1000 cyclic calculations using a cross-validation likelihood (CVL) algorithm. Based on the regression coefficient of each lncRNA in the optimal lncRNA combination, lncRNA expression weighted by these coefficients was used to establish the risk prediction model, from which the risk score (RS) of each sample was obtained. The RS calculation formula was as follows:
Validation of the risk prediction model
In order to validate the risk prediction model, GSE32062 and GSE17260 were used as single validation sets. Datasets meeting the following criteria were selected as independent validation datasets: 1) all data contained gene expression profiles; 2) detection objects were solid tumour tissues from patients with OC, exclusive of blood and cell lines; 3) expression profiles were all from human subjects; 4) samples had prognostic clinical information and 5) the sample number was no less than 50. This led to the selection of the three microarray datasets GSE49997, GSE26712, and GSE31245 containing 204, 185, and 58 samples, respectively. We also downloaded the mRNA-seq expression profiles of cervical cancer (CESC) samples with the corresponding clinical information data from TCGA to further validate the efficiency of the risk prediction model. Of 309 samples that were downloaded, 293 had clinical information available and were therefore used for validation. The risk-score model was used to assess significant prognostic differences between high and low risk groups, with the results determining the stability of this established model.
Analysis of important lncRNA relevant pathways
Gene sets were isolated from the modules that contained the optimal lncRNAs significantly associated with OC prognosis. The GSEA (gene set enrichment analysis) method [28] (http://software.broadinstitute.org/gsea/index.jsp) was then used to identify KEGG pathways enriched in these optimal lncRNA-related gene sets. The GSEA-based pathway enrichment analysis was performed on each lncRNA counter-gene in the lncRNA-mRNA network. Three key statistical values were used in this analysis. The first of these, the enrichment score (ES), is the original result of the GSEA analysis, reflecting the degree of enrichment of one functional gene set locating at the anterior or posterior of this gene sequence after all the hybridization data are ordered. The fundamental calculation principle is to scan the collating sequence; when a gene of this set is prepared, the ES value will increase, otherwise it will decrease. The second key statistical value is the normalised enrichment score (NES) that results from standardized processing of ES values. The final statistical value is the nominal P value, which describes the statistical significance of the enrichment scores of a functional gene subset; a smaller P value indicates a better degree of gene enrichment. When the NES absolute value increases, the P value will spontaneously decrease, suggesting a higher degree of enrichment and a higher significance of the result. In this study, a P value less than 0.05 was chosen as the threshold to screen KEGG pathways that were significantly enriched in the relevant module genes.
Results
Screening of stable modules significantly correlated with OC using WGCNA
One dataset from TCGA and two datasets from NCBI GEO (GSE32062 and GSE17260) containing RNA expression data for OC were used for analysis. We identified 13,251 mRNAs and 646 lncRNAs overlapping in the three datasets (TCGA, GSE32062, and GSE17260). TCGA expression data was used as the training set and the remaining two datasets were regarded as validation datasets. First, the consistency of the expression values of the overlapped RNA in the three datasets was checked. The results indicated that the correlation distribution of RNA expression levels between each pair of datasets was higher than 0.9 with P values less than 1e− 200, suggesting that all three sets displayed significant positive correlation (Fig. 1).

Correlation of expression levels between each pair of the three datasets; TCGA, GSE32062 and GSE17260. The graphs (from left to right) show the comparisons: TCGA-GSE32062 (a), TCGA-GSE17260 (b) and GSE32062-GSE17260 (c)
WGCNA analysis was further conducted to screen stable modules associated with OC. The β (power) value of 3 was chosen as this was its value when R^2 was approximately 0.9 for the first time (Fig. 2a). This value of the β parameter not only ensured that the network connection was close to the scale-free distribution, but was also the minimum threshold to give the curve a tendency to be smooth. When the β value was equal to 3, the RNA average connection degree in the network was 3 (Fig. 2b), conforming to the small-world network character in scale-independent networks.

Selection graphs of the weight parameter β in the adjacency matrix (a) and schematic diagram of the average connectivity of RNA under various power parameters (b). The horizontal and vertical axes represent the weight parameter power and the square values (R^2) of correlation coefficients between log (k) and log (p(k)) in the corresponding network, respectively. The red line represents the standard line when R^2 reached a value of 0.9
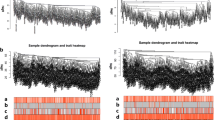
An RNA adjacency matrix was constructed and a hierarchical clustering tree was built on basis of this matrix. Based on the criteria of a cut height equal to 0.99 and an involvement of at least 200 RNAs per module, six modules were identified; M1-blue, M2-brown, M3-green, M4-grey, M5-turquoise, and M6-yellow (Fig. 3a). The corresponding modules were partitioned in the single validation datasets GSE32062 (Fig. 3b) and GSE17260 (Fig. 3c), based on their inclusive RNA in the training set (TCGA) to evaluate the stability of modules selected from the training set. The results of the partition and correlation of modules in the TCGA dataset are displayed in Fig. 4. It was found that RNA in the same modules (i.e. dots with same colour) tended to aggregate together (Fig. 4a), indicating that RNA in the same module had similar expression. The clustering results of module RNA in the other two datasets indicated that the blue, brown and green modules exhibited characteristics of independent branches (Fig. 4b).

The tree views of RNA module division in the datasets TCGA (a), GSE32062 (b), and GSE17260 (c). Each colour represents one module

Multidimensional expansion map (MDS) of each gene located in a module of the TCGA dataset (a), and system cluster tree diagram of modules in datasets GSE32062 and GSE17260 (b). The X and Y axes represent the matching first and second principal components, respectively
The results of stability analysis on the above six modules showed that the stability scores (preservation Z-score) of the blue, yellow, brown and green modules were all higher than 5, whereas those for the gray and turquoise modules were not. RNAs included in these four stable functional modules were likely associated with OC pathogenesis. Table 1 lists the functional annotation information of the six modules, showing that RNAs in the top four highly stable modules were mainly involved in cellular immune response, cell adhesion, sexual reproduction, and the cell cycle.
The corresponding clinical information of samples in the TCGA dataset was integrated and the correlation between RNA obtained by partitioning in each module and clinical factors was calculated (Fig. 5). The results showed that the four highly stable RNA modules brown, yellow, blue and green were significantly correlated using OC data including stage, grade, radiotherapy, lymph node metastasis, and recurrence. Therefore, a total of 33 lncRNAs in the four highly stable modules which were enriched for specific biological functions and displayed significant relationships with clinical factors of OC were selected as important OC-related factors for subsequent analyses.

Correlation analyses between functional modules of RNAs in the TCGA dataset and their clinical attributes
Establishment and evaluation of risk assessment model
Selection of optimal lncRNA combinations
We used Cox univariate regression analysis (with the regression threshold P set at 0.05) to evaluate the expression levels of the 33 lncRNAs within the stable modules of the TCGA dataset with regard to the OC sample clinical prognosis information. This resulted in the identification of 19 prognostic related-lncRNAs (Table 2).
Next, we optimized and screened the 19 OC prognostic-related lncRNAs using a Cox-PH model based on an L1-penalised regularization regression algorithm in the penalised package, in which the value of the “lambda” parameter was obtained by 1000 cyclic calculations using a CVL algorithm. This resulted in an optimal prognosis combination containing 5 lncRNAs: GAS5, HCP5, PART1, SNHG11 and SNHG5 (Table 3).
Construction, evaluation and selection of risk prediction models
Based on the corresponding Cox-PH regression coefficients of the optimal combination of 5 lncRNAs obtained in the previous section, the prediction model of sample risk scoring was established as follows:
The RS of each sample in the training set (TCGA) was calculated according to the constructed model. These samples were divided into high- and low-risk groups based on the scoring medians. Next, Kaplan-Meier survival curve analysis was used to examine the significant differences in survival time between the two sample groups. The result indicated that this model had an excellent distinguishing effect on the sample groups (p value < 0.01) (Fig. 6 and Fig. 7); therefore we used this model for subsequent analyses.

Kaplan-Meier survival curves of samples in training set TCGA (a), validation sets GSE32062 (b) and GSE17260 (c), and independent validation sets GSE26712 (d), GSE31245 (e), and GSE49997 (f) using the risk assessment model established by the 5 optimal lncRNAs. The green and red curves represent the low- and high-risk groups, respectively

Kaplan-Meier survival curves of samples in validation set of cervical cancer from TCGA to validate the efficiency of the risk prediction model
As shown in Fig. 7, the prediction model also displayed significant differentiation effects on samples of the GSE32062 and GSE17260 datasets included in the WGCNA analysis (P value < 0.05). The stability of this model was further validated by the independent validation datasets GSE49997, GSE26712, and GSE31245, as well as the CESC data from TCGA exclusive of any processing (including module discovering and survival analysis). The model displayed significant differentiation effects for all these validation sets, indicating a high robustness of this prediction model. Taken together, the above results show that the 5 lncRNAs comprising the key components of this risk prediction model (GAS5, HCP5, PART1, SNHG11, and SNHG5), were significantly associated with OC prognosis, and form a stable combination for distinguishing high- and low-risk prognostic samples, as well as other unprocessed samples.
Network construction and functional pathway analysis
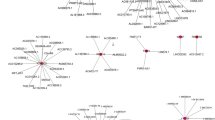
The previous analysis identified the 5 important OC-related lncRNAs; GAS5, HCP5, PART1, SNHG11, and SNHG5. These five lncRNAs were distributed in the blue, green, and yellow stable modules discovered by the WGCNA method. The mRNAs closely related to the expression of these five lncRNAs can be found in these modules, and may be the target genes of these key lncRNAs (Fig. 8).

Graphs of lncRNA-mRNA networks corresponding to the blue, green, and yellow modules. The circular node represents mRNA, and the square node represents lncRNA. The colour of the node indicates that it belongs to the corresponding module, and the green and red connecting lines indicate negative and positive correlation, respectively
GSEA was used to perform pathway enrichment analysis of these mRNAs. This showed that the network modules related to the 5 prognostic genes were significantly associated with cell local adhesion, cancer signalling pathways, JAK-STAT signalling, and endogenous cell receptor interaction (Table 4).
Discussion
OC is the most deadly gynaecological cancer and a primary cause of female cancer-related deaths worldwide, yet the regulatory machinery underlying OC development remains unclear. Increasing evidence has identified lncRNAs in multiple biological functions at various stages of OC development, and deregulated expression of lncRNA is closely associated with OC early diagnosis, prognosis, and response to chemotherapy [4,5,6,7,8,9]. To identify a novel lncRNA-based signature for predicting prognosis of OC patients, RNA expression profiling data and clinical survival prognosis information from a large number of OC patients were downloaded from the public database. A co-expression network was subsequently constructed and modules with OC-related biological functions were excavated.
As a bioinformatics algorithm for construction of co-expression networks, WGCNA is commonly used to identify modules associated with diseases and consequently screen important pathogenic mechanisms or potential therapeutic targets [24]. So far, gene modules associated with several cancers have been identified and validated through co-expression network analysis [29,30,31]. In the present study, six modules were obtained in the training set, four of which (blue, yellow, brown, and green) displayed high stability. The results of functional annotation analysis showed that RNAs in these four highly stable modules were mainly involved in cellular immune responses, cell adhesion, the cell cycle, and sexual reproduction, indicating their likely association with OC pathogenesis.
Prognostic genes are informative for cancer prognosis and treatment because of their potential as biomarkers, and can help to predict patients’ survival, as well as providing insights into the molecular mechanisms of tumour progression [31,32,33,34,35]. In the present study, WGCNA analysis identified a total of 33 lncRNAs in the four stable modules with relevant biological functions and correlation with specific clinical factors of OC. Based on the expression level of these 33 lncRNAs in the TCGA dataset and the clinical prognostic information of the samples, a short-list of 19 lncRNAs were screened using univariate Cox regression analysis. Finally, an optimal prognosis combination containing 5 lncRNAs (GAS5, HCP5, PART1, SNHG11, and SNHG5) was identified using a Cox-PH model. Considering that risk assessment tools can help to detect high-risk populations for a disease [15], the present study establishes a new risk assessment system based on the above prognostic gene signature. The effectiveness of the RS model was tested in both the training set and validation sets, and the results indicated that the risk assessment tool could successfully distinguish a population at high risk of future OC development. This method is simple and inexpensive enough to be used in normal clinical practice and mass screening. Compared with the previous lncRNA signatures and RS models for OC [3, 6, 9, 19], the present prognostic lncRNA combination and RS model may be more reliable because they were screened based on the WGCNA analysis instead of single differential gene analysis.
The five prognostic genes have all been reported to be associated with human cancer, and three (GAS5, HCP5, and SNHG11) have reported associations with OC. GAS5 (growth arrest-specific transcript 5) was originally isolated from NIH 3 T3 cells using subtraction hybridization [36]. The latest studies demonstrated that GAS5 usually functions as a tumour suppressor to control apoptosis of various cancer cells, including breast cancer, prostate cancer, renal cell carcinoma, and ovarian cancer [12, 37,38,39]. Furthermore, GAS5 acts as tumour suppressor and has been suggested as a potential target for diagnosis and therapy of OC [12]. HCP5 (HLA Class I Histocompatibility Antigen Protein P5) is localised in the major histocompatibility complex (MHC) class I region and has involvement in the development of various tumours including OC [40,41,42,43]. SNHG11 (small nucleolar RNA host gene 11) is an obesity-associated lncRNA, and is involved in positive regulation of cell proliferation in OC [44]. PART-1 (prostate androgen regulated transcript 1) is a gene known to be predominantly expressed in the prostate- and androgen-regulated. Its aberrant expression has been associated with poor prognosis of prostate cancer, non-small cell lung cancer, and colorectal cancer, leading to the suggestion of its use as a novel tumour marker [45,46,47]. SNHG5 (small nucleolar RNA host gene 5) has been strongly implicated in cancer-related processes, such as cell differentiation, cell proliferation, and metastasis [48,49,50]. The strong evidence of the association of all five lncRNAs with cancer and/or OC supports the conclusion that this study identified potential biomarkers for predicting the prognosis of OC patients, which should also help future research into the pathogenesis of OC.
It has been demonstrated that lncRNAs play important roles in a variety of biological processes by regulating target genes at transcriptional, posttranscriptional and epigenetic levels [51, 52]. Therefore, we investigated the target genes regulated by the 5 prognostic lncRNAs to decipher their potential biological function in the pathogenesis of OC. The result of pathway enrichment analysis showed that the network modules related to the five prognostic genes were significantly associated with cell local adhesion, cancer signalling pathways, JAK-STAT signalling, and endogenous cell receptor interaction. According to functional analyses of lncRNA regulators, it was found that low expression of GAS5 could promote proliferation, metastasis, and infiltration of OC cells, and as a result, was considered to be associated with poor prognosis of OC [11, 12]. Most of the mRNAs in the regulated pathway are associated with the development of OC. For example, up-regulation of RASSF5 expression can inhibit the growth of OC cells [53], over-expression of FZD3 can increase the survival time of OC patients [54], and inhibition of MMP9 gene expression can block metastasis of ovarian cancer cells [55]. Therefore, the five prognostic-related lncRNAs identified in the present study may play roles in the initiation and development of OC by regulating genes involved in cell adhesion and the JAK-STAT signalling pathway.
Although the independent validation performed in this study and the results of previous reports both indicate that the present model should be effective, there are limitations of the present study. Primarily, as this was an extensive bioinformatics study based on previously published data, our results should be further validated using in vitro and in vivo models. However, our results form a strong basis for other researchers to carry out the relevant future research.
Conclusions
In conclusion, our study constructed a co-expression network and excavated four modules with specific biological functions related to OC. A risk assessment tool for predicting prognosis of OC was further identified and validated based on the expression of 5 prognostic genes. The present risk assessment tool could provide a novel reliable method to identify individuals at high risk of OC, and the 5 prognostic genes could be promising prognostic biomarkers for OC.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from TCGA database (https://gdc-portal.nci.nih.gov/) and GEO database (http://www.ncbi.nlm.nih.gov/geo/) with assession number of GSE32062 [56] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32062), GSE17260 [57] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17260), GSE49997 [58] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49997), GSE26712 [59, 60] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26712) and GSE31245 [61] (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31245). Processed data are available from the corresponding author on reasonable request.
Abbreviations
- CESC:
-
Cervical cancer
- CVL:
-
Cross-validation likelihood
- ES:
-
Enrichment score
- GSEA:
-
Gene set enrichment analysis
- lncRNAs:
-
Long non-coding RNAs
- NES:
-
Normalized enrichment score
- OC:
-
Ovarian cancer
- RS:
-
Risk score
- WGCNA:
-
Weighted gene co-expression network analysis
References
Siegel R, Ma JM, Zou ZH, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29.
Yang K, Hou Y, Li A, Li ZZ, Wang WJ, Xie HY, Rong ZW, Lou G, Li K. Identification of a six-lncRNA signature associated with recurrence of ovarian cancer. Sci Rep. 2017;7(1):752.
Rustin G, dBM V, Griffin C, Qian W, Swart AM. Early versus delayed treatment of relapsed ovarian cancer. Lancet. 2010;376(9747):1155–63.
Tripathi MK, Doxtater K, Keramatnia F, Zacheaus C, Yallapu MM, Jaggi M, Chauhan SC. Role of lncRNAs in ovarian cancer: defining new biomarkers for therapeutic purposes. Drug Discov Today. 2018;23(9):1635–43.
Cheng ZP, Guo J, Chen L, Luo N, Yang WH, Qu XY. A long noncoding RNA AB073614 promotes tumorigenesis and predicts poor prognosis in ovarian cancer. Oncotarget. 2015;6(28):25381–9.
Liu R, Zeng Y, Zhou CF, Wang Y, Li X, Liu ZQ, Chen XP, Zhang W, Zhou HH. Long noncoding RNA expression signature to predict platinum-based chemotherapeutic sensitivity of ovarian cancer patients. Sci Rep. 2017;7(1):18.
Ren CC, Li XB, Wang TZ, Wang GY, Zhao C, Liang T, Zhu YY, Li MH, Yang C, Zhao YL, et al. Functions and mechanisms of long noncoding RNAs in ovarian cancer. Int J Gynecol Cancer. 2015;25(4):566–9.
Yim GW, Kim HJ, Kim LK, Kim SW, Kim S, Nam EJ, Kim YT. Long non-coding RNA HOXA11 antisense promotes cell proliferation and invasion and predicts patient prognosis in serous ovarian cancer. Cancer Res Treat. 2017;49(3):656–68.
Zhan XH, Dong CP, Liu G, Li YX, Liu L. Panel of seven long noncoding RNA as a candidate prognostic biomarker for ovarian cancer. Onco Targets Ther. 2017;10:2805–13.
Chen ZJ, Zhang Z, Xie BB, Zhang HY. Clinical significance of up-regulated lncRNA NEAT1 in prognosis of ovarian cancer. Eur Rev Med Pharmacol Sci. 2016;20(16):3373–7.
Li J, Huang H, Li YG, Li L, Hou WH, You ZS. Decreased expression of long non-coding RNA GAS5 promotes cell proliferation, migration and invasion, and indicates a poor prognosis in ovarian cancer. Oncol Rep. 2016;36(6):3241–50.
Li J, Yang C, Li YG, Chen AY, Li L, You ZS. LncRNA GAS5 suppresses ovarian cancer by inducing inflammasome formation. Biosci Rep. 2017;38(2):BSR20171150.
Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322(5903):881–8.
Chen JM, Cooper DN, Chuzhanova N, Férec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8(10):762–75.
Iida M, Ikeda F, Hata J, Hirakawa Y, Ohara T, Mukai N, Yoshida D, Yonemoto K, Esaki M, Kitazono T. Development and validation of a risk assessment tool for gastric cancer in a general Japanese population. Gastric Cancer. 2018;21(3):383–90.
Pavlou M, Ambler G, Seaman SR, Guttmann O, Elliott P, King M, Omar RZ. How to develop a more accurate risk prediction model when there are few events. BMJ. 2015;351:h3868.
Hung YC, Lin CL, Liu CJ, Hung H, Lin SM, Lee SD, Chen PJ, Chuang SC, Yu MW. Development of risk scoring system for stratifying population for hepatocellular carcinoma screening. Hepatology. 2015;61(6):1934–44.
Hussein AA, Ghani KR, Peabody J, Sarle R, Abaza R, Eun D, Hu J, Fumo M, Lane B, Montgomery JS, et al. Development and validation of an objective scoring tool for robot-assisted radical prostatectomy: prostatectomy assessment and competency evaluation. J Urol. 2017;197(5):1237–44.
Zhou M, Sun YY, Sun YF, Xu WY, Zhang ZY, Zhao HQ, Zhong ZH, Sun J. Comprehensive analysis of lncRNA expression profiles reveals a novel lncRNA signature to discriminate nonequivalent outcomes in patients with ovarian cancer. Oncotarget. 2016;7(22):32433–48.
Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93.
Larkin MA, Blackshields G, Brown NP, Chenna R, Mcgettigan PA, Mcwilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8.
Parrish RS. Effect of normalization on significance testing for oligonucleotide microarrays. J Biopharm Stat. 2004;14(3):575–89.
Ritchie ME, Phipson B, Wu D, Hu YF, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Zhai XF, Xue QF, Liu Q, Guo YY, Chen Z. Colon cancer recurrence-associated genes revealed by WGCNA co-expression network analysis. Mol Med Rep. 2017;16(5).
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559.
Tibshirani R. The lasso method for variable selection in the cox model. Stat Med. 1997;16(4):385–95.
Goeman JJ. L1 penalized estimation in the cox proportional hazards model. Biom J. 2010;52(1):70–84.
Subramanian A, Tamayo P, K MV, S M, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50.
Chou WC, Cheng AL, Brotto M, Chuang CY. Visual gene-network analysis reveals the cancer gene co-expression in human endometrial cancer. BMC Genomics. 2014;15(1):300.
Liu R, Cheng Y, Yu J, Lv QL, Zhou HH. Identification and validation of gene module associated with lung cancer through coexpression network analysis. Gene. 2015;563(1):56–62.
Yang Y, Han L, Yuan Y, Li J, Hei NN, Liang H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun. 2014;5:3231.
Adler AS, Chang HY. From description to causality: mechanisms of gene expression signatures in cancer. Cell Cycle. 2006;5(11):1148–51.
Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98(4):262–72.
Spentzos D, Levine DA, Ramoni MF, Joseph M, Gu XS, Boyd J, Libermann TA, Cannistra SA. Gene expression signature with independent prognostic significance in epithelial ovarian cancer. J Clin Oncol. 2004;22(23):4700–10.
Zhao HJ, Ljungberg B, Grankvist K, Rasmuson T, Tibshirani R, Brooks JD. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 2005;3(1):e13.
Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54(6):787–93.
Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28(2):195–208.
Pickard MR, Mourtada-Maarabouni M, ., Williams GT: Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim Biophys Acta (BBA) - Mol Basis Dis 2013, 1832(10):1613–1623.
Qiao HP, Gao WS, Huo JX, Yang ZS. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac J Cancer Prev. 2013;14(2):1077–82.
Chen X, Guo W, Xu XJ, Su F, Wang Y, Zhang Y, Wang Q, Zhu L. Melanoma long non-coding RNA signature predicts prognostic survival and directs clinical risk-specific treatments. J Dermatol Sci. 2017;85(3):226–34.
Liang LL, Xu JC, Wang M, Xu GR, Zhang N, Wang GZ, Zhao YF. LncRNA HCP5 promotes follicular thyroid carcinoma progression via miRNAs sponge. Cell Death Dis. 2018;9(3):372.
Liu N, Zhang R, Zhao XM, Jiaming SU, Bian XL, Jinsong NI, Yue Y, Cai Y, Jin JJ. A potential diagnostic marker for ovarian cancer: involvement of the histone acetyltransferase, human males absent on the first. Oncol Lett. 2013;6(2):393–400.
Yu Y, Shen HM, Fang DM, Meng QJ, Xin YH. LncRNA HCP5 promotes the development of cervical cancer by regulating MACC1 via suppression of microRNA-15a. Eur Rev Med Pharmacol Sci. 2018;22(15):4812–9.
Li YS, Chen J, Zhang JW, Wang ZS, Shao TT, Jiang CJ, Xu J, Li X. Construction and analysis of lncRNA-lncRNA synergistic networks to reveal clinically relevant lncRNAs in cancer. Oncotarget. 2015;6(28):25003–16.
Hu YB, Ma Z, He YM, Liu W, Su Y, Tang ZB. PART-1 functions as a competitive endogenous RNA for promoting tumor progression by sponging miR-143 in colorectal cancer. Biochem Biophys Res Commun. 2017;490(2):317–23.
Li M, Zhang WW, Zhang SJ, Wang CH, Lin YP. PART1 expression is associated with poor prognosis and tumor recurrence in stage I-III non-small cell lung cancer. J Cancer. 2017;8(10):1795–800.
Sidiropoulos M, Chang A, Jung K, Diamandis EP. Expression and regulation of prostate androgen regulated transcript-1 (PART-1) and identification of differential expression in prostatic cancer. Br J Cancer. 2001;85(3):393–7.
Ichigozaki Y, Fukushima S, Jinnin M, Miyashita A, Nakahara S, Tokuzumi A, Yamashita J, Kajihara I, Aoi J, Masuguchi S, et al. Serum long non-coding RNA, snoRNA host gene 5 level as a new tumor marker of malignant melanoma. Exp Dermatol. 2016;25(1):67–9.
Shi J, Xu M, Tang Q, Zhao K, Deng A, Li J. Highly sensitive determination of diclofenac based on resin beads and a novel polyclonal antibody by using flow injection chemiluminescence competitive immunoassay. Spectrochim Acta A Mol Biomol Spectrosc. 2018;191:1–7.
Shen HJ, Wang Y, Shi WD, Sun GX, Hong LJ, Zhang Y. LncRNA SNHG5/miR-26a/SOX2 signal axis enhances proliferation of chondrocyte in osteoarthritis. Acta Biochim Biophys Sin. 2018;50(2):191–8.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11(1):59.
Li BT, Yu C, Xu Y, Liu SB, Fan HY, Pan WW. TET1 inhibits cell proliferation by inducing RASSF5 expression. Oncotarget. 2017;8(49):86395–409.
Seagle BL, Dandapani M, Yeh JY, Shahabi S. Wnt signaling and survival of women with high-grade serous ovarian cancer: a brief report. Int J Gynecol Cancer. 2016;26(6):1078–80.
Liu H, Zeng Z, Wang S, Li T, Mastriani E, Li QH, Bao HX, Zhou YJ, Wang X, Liu Y. Main components of pomegranate, ellagic acid and luteolin, inhibit metastasis of ovarian cancer by down-regulating MMP2 and MMP9. Cancer Biol Ther. 2017;18(12):990–9.
Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Fujiwara H, Masuzaki H, Katabuchi H, Kawakami Y, Okamoto A, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18(5):1374–85.
Yoshihara K, Tajima A, Yahata T, Kodama S, Fujiwara H, Suzuki M, Onishi Y, Hatae M, Sueyoshi K, Fujiwara H, et al. Gene expression profile for predicting survival in advanced-stage serous ovarian cancer across two independent datasets. PLoS One. 2010;5(3):e9615.
Pils D, Hager G, Tong D, Aust S, Heinze G, Kohl M, Schuster E, Wolf A, Sehouli J, Braicu I, et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: a study of the OVCAD consortium. Cancer Sci. 2012;103(7):1334–41.
Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, Ozbun L, Brady J, Barrett JC, Boyd J, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68(13):5478–86.
Vathipadiekal V, Wang V, Wei W, Waldron L, Drapkin R, Gillette M, Skates S, Birrer M. Creation of a human Secretome: a novel composite library of human secreted proteins: validation using ovarian Cancer gene expression data and a virtual Secretome Array. Clin Cancer Res. 2015;21(21):4960–9.
Spentzos D, Levine DA, Kolia S, Otu H, Boyd J, Libermann TA, Cannistra SA. Unique gene expression profile based on pathologic response in epithelial ovarian cancer. J Clin Oncol. 2005;23(31):7911–8.
Acknowledgements
Not applicable.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
ZQ conceived and designed the study, performed data analyses and wrote the manuscript. FCH contributed significantly to both data analyses and manuscript revision. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhao, Q., Fan, C. A novel risk score system for assessment of ovarian cancer based on co-expression network analysis and expression level of five lncRNAs. BMC Med Genet 20, 103 (2019). https://doi.org/10.1186/s12881-019-0832-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0832-9