
Abstract
Background
WNT1 mutations cause bone fragility as well as brain anomalies. There are some reported cases of WNT1 mutations with normal cognition. Genotype and phenotype correlation of WNT1 mutations has not been established.
Case presentation
Here we present two female siblings with osteogenesis imperfecta (OI) born to a consanguineous couple. Both sustained severe bone deformities. However, only the younger had severe brain anomalies resulting in an early death from pneumonia, while the older had normal intellectual development. Next generation sequencing showed a homozygous mutation, c.6delG, p.Leu3Serfs*36 in WNT1. To our knowledge, it is the most 5′ truncating mutation to date.
Conclusion
This report emphasizes the intrafamilial variability of brain anomalies found in this OI type and suggests that WNT1 may not be necessary for normal human cognitive development.
Similar content being viewed by others

Background
Osteogenesis imperfecta (OI) is a heritable connective tissue disease characterized by bone fragility and fracture susceptibility. More than 95% of OI have dominant mutations in COL1A1 or COL1A2, resulting in primary structural or quantitative defects of collagen type I. The recessive forms of OI are caused by mutations in CRTAP, BMP1, CREB3L1, IFITM5, FKBP10, LEPRE1, PPIB, SP7, PLOD2, TMEM38B, SERPINF, SERPINH1, SEC24D, SPARC, WNT1 [1], which lead to defects of proteins interacting with collagen post-translationally. Recently, an X-linked form caused by mutations in MBTPS2 was identified [2].
OI has been classified into several types according to clinical features and genetic alterations. Type XV OI, first described by Keupp in 2013, is caused by biallelic mutations in WNT1 [3]. So far, at least 30 cases have been reported. In addition to bone fragility, many of them had neurological abnormalities [4,5,6,7]. Defects in Wnts or related proteins could lead to derangements in axonal pathways, including commissural axon tracts such as the corpus callosum [8]. Monoallelic mutations in WNT1 cause early onset osteoporosis [9]. Genotype and phenotype correlation of WNT1 mutations is not yet determined, requiring a larger cohort of patients.
Here, we describe two siblings with OI. One of them had brain malformations and global developmental delay, while the other had normal cognition. Mutation analysis identified a homozygous frameshift mutation in WNT1, which is the most 5′ change reported to date.
Case presentation
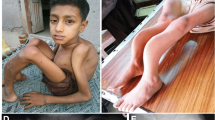
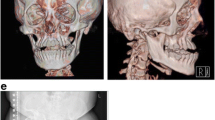
A 14-year-old Thai girl was born via cesarean section due to premature rupture of the membrane with a birth weight of 2500 g. She is the first child of a consanguineous (second-degree relatives) couple. Both parents are healthy and have never had fractures. During her first year of life, she had delayed motor development and growth failure. At one year of age, she could not sit by herself and weighed 7.5 kg (< 3rd centile). She presented to our hospital at 14 months of age with fractures of both femora without a history of significant trauma. She was found to have ptosis of both eyes with normal teeth but no blue sclerae. She was small for her age. Her weight was 7.8 kg (3rd centile) and her length was 68 cm (< 3rd centile). Skeletal survey showed diffuse osteopenia, multiple healed fractures of the right humoral shaft, both tibiae and fibulae. Spine radiograph showed flattening and indentation of vertebral bodies (Fig. 1). A diagnosis of OI was made and intravenous bisphosphonate therapy (pamidronate 1 mg/kg/dose for 3 days) was initiated and given every 3 months. However, she sustained 1–2 long bone fractures per year from minor trauma. She required multiple corrective osteotomies to correct her deformities. At the last follow-up, she was 14 years old, weighing 20 kg. She could not walk due to her long bone deformity (Fig. 1). Remarkably, although she was in a special education class due to physical disabilities, her cognition was appropriate for age. She could talk fluently and do mathematics properly.

Radiological features of the proband. Imaging of the thoracic and lumbar spines at 14 months of age, a the antero-posterior and b lateral views revealed depressed multiple vertebrae. Figures c-f showed imaging at 14 years of age of upper extremities (c-d) and lower extremities (e-f) revealing deformities of humeri, left ulna and radius, right tibia and fibula, left tibia and fibula, respectively
Prenatally, her younger sister was found to have a dilated fourth ventricle by an ultrasonography. She was born at term via cesarean section because of previous cesarean section and was diagnosed with hydrocephalus at birth. At 4 months of age, she had her first fracture without a history of a significant trauma, leading to a diagnosis of OI. Physical examination revealed a head circumference of 38 cm (> 95th centile) with a wide anterior fontanelle (3 × 3 cm.) and blue sclerae. She had global developmental delay (could not hold her head) and hypotonia. MRI of the brain demonstrated a large posterior fossa cyst connecting with the fourth ventricular system, moderate hydrocephalus, hypoplasia of cerebellar hemisphere with absence of cerebellar vermis, and hypoplasia of corpus collosum. She was also diagnosed with vesicoureteral reflux grade V and gastroesophageal reflux requiring tube feeding. The patient had multiple hospitalizations because of recurrent urinary tract infections and pneumonia. She expired at the age of one year.
Sixteen known OI genes, BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, SP7, TMEM38B, WNT1, and MBTPS2, were amplified from 200 ng of genomic DNA using the Truseq Custom Amplicon Sequencing kit (Illumina, San Diego, CA). 286 amplicons which covered all the 226 exons (28 kb) of the target genes were sequenced by Miseq (Illumina, San Diego, CA) using 2 × 250 paired-end reads. SNVs and Indels were detected by Miseq reporter software. The proband was found to harbor a homozygous mutation, c.6delG, p.Leu3Serfs*36 in WNT1. The mutation has never been reported in Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php) (Fig. 2). The mutation was subsequently confirmed by PCR-Sanger sequencing. Segregation analysis was performed by using primers, WNT1-E1F: GGT TGTTAAAGCCAGACTGC and WNT1-E1R: ACCAGCTCACTTACCACCAT. The results revealed that the patient was homozygous, while her mother was heterozygous for the mutation (Fig. 3).


Mutation analysis. Sanger sequencing shows that the proband is homozygous while his mother is heterozygous for the WNT1 c.6delG, p.Leu3Serfs*36 mutation
Discussion and conclusions
To better understand the clinical manifestations and natural history of patients with type XV OI, more patients and long-term follow-up are needed. Here, we report two siblings with a WNT1 mutation. The older and younger sisters had their first fractures at 14 and four months of ages, respectively. Despite regular pamidronate administration for the older sister starting since then, at her last follow-up at 14 years of age, she sustained severe deformities of all extremities. The natural history of first fractures after birth, but severe bone deformities later in life is similar to the previous reported cases with WNT1 mutations (Table 1: 55%, 10/18). Many OI patients with COL1A1 or COL1A2 mutations had fractures prenatally but did not have such severe bone deformities in their teens. This emphasizes the fact that onsets of fractures in patients with OI do not correlate with severities of final bone deformities.
Some other features of OI are inconsistent between our two patients. Blue sclerae were observed in only the younger sister, but not the proband. Previous reports found that blue sclerae could be observed in some cases with WNT1 mutations (Table 1). Consistent with previous studies, neither of our patients had dentinogenesis imperfecta. Notably, ptosis, one of the most common clinical presentations of OI patients with WNT1 mutations was only found in the proband. It is not usually found in other types of OI. Brain anomalies or intellectual disabilities were observed in 33% (9/27) of the previous reported cases (Table 1). Many patients have normal cognitive development. Remarkably, our proband had normal intellectual development, while her younger sister had severe brain anomalies, including hydrocephalus detected prenatally. This demonstrates a significant variability in neurological involvement between the affected siblings in this family and suggests that neurological abnormalities in WNT1 mutations might be subject to modifier genes, epigenetics or non-genetic factors. There are two other families in the literature with significantly different neurological manifestations among the family members; one affected sib had normal intelligence and the other had severe brain anomalies or delayed development [5, 6]. Intrafamilial variable expression of this gene was substantiated.
Using Truseq Custom Amplicon sequencing, our proband was found to have an autosomal recessive form of OI caused by a homozygous truncating mutation in WNT1. Her sister’s blood sample could not be obtained. With similar recurrent bone fractures, we assumed that she harbored the same homozygous mutation. WNT1 mutations were identified as a cause of malformations of midbrain and cerebellum in early brain development in mice long before it was identified as a cause of OI in humans [10]. In 2014, Swaying (Wnt1sw/sw) mice carrying WNT1 mutations were found to have OI phenotypes including bone fragility and severe osteopenia [4]. The first report of a patient with a WNT1 mutation was in 2013 [3]. As far as we know, the mutation identified in our patient is the first reported mutation in exon 1 and is the most 5′ truncating mutation (Fig. 2). The out-of-frame G deletion at the starting codon of WNT1 is expected to result in a nonsense-mediated mRNA decay (NMD). There are no in-frame methionines after the frameshifting variant, so there would not be the possibility of another in-frame start site.
The genotype and phenotype correlation of WNT1 mutations has not been established. The homozygous mutation found in our proband, which is a frameshift starting from the second codon, is expected to lead to a nonfunctional WNT1. She had normal cognition at the age of 14 years, indicating that humans without functional WNT1 could be intellectually normal.
In summary, we report two siblings with an autosomal recessive OI caused by the most 5′ homozygous mutation in WNT1. The findings exemplify intrafamilial variability in the neurological phenotype and suggest that WNT1 may not be necessary for normal human cognitive development.
Abbreviations
- COL1A1:
-
Collagen type I, alpha 1
- COL1A2:
-
Collagen, type I, alpha 2
- HGMD:
-
Human Gene Mutation Database
- MRI:
-
Magnetic resonance imaging
- OI:
-
Osteogenesis imperfecta
- PCR:
-
Polymerase chain reaction
- WNT1:
-
Wnt Family Member 1
References
Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, Montpetit K, Semler O. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052.
Lindert U, Cabral WA, Ausavarat S, Tongkobpetch S, Ludin K, Barnes AM, Yeetong P, Weis M, Krabichler B, Srichomthong C, Makareeva EN, Janecke AR, Leikin S, Röthlisberger B, Rohrbach M, Kennerknecht I, Eyre DR, Suphapeetiporn K, Giunta C, Marini JC, Shotelersuk V. MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun. 2016; https://doi.org/10.1038/ncomms11920.
Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Janda CY, Becker J, Breer S, Altunoglu U, Grünhagen J, Krawitz P, Hecht J, Schinke T, Makareeva E, Lausch E, Cankaya T, Caparrós-Martín JA, Lapunzina P, Temtamy S, Aglan M, Zabel B, Eysel P, Koerber F, Leikin S, Garcia KC, Netzer C, Schönau E, Ruiz-Perez VL, Mundlos S, Amling M, Kornak U, Marini J, Wollnik B. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92:565–74.
Aldinger KA, Mendelsohn NJ, Chung BH, Zhang W, Cohn DH, Fernandez B, Alkuraya FS, Dobyns WB, Curry CJ. Variable brain phenotype primarily affects the brainstem and cerebellum in patients with osteogenesis imperfecta caused by recessive WNT1 mutations. J Med Genet. 2016;53:427–30.
Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, Fernandez BA, Elsayed SM, Elsobky E, Verma I, Nair S, Turner EH, Smith JD, Jarvik GP, Byers PH. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013;92:590–7.
Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, Nieminen-Pihala V, Aronen M, Laine T, Kröger H, Cole WG, Lehesjoki AE, Nevarez L, Krakow D, Curry CJ, Cohn DH, Gibbs RA, Lee BH, Mäkitie O. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368:1809–16.
Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. J Med Genet. 2013;50:491–2.
Freese JL, Pino D, Pleasure SJ. Wnt signaling in development and disease. Neurobiol Dis. 2010;38:148–53.
Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50:345–8.
McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–85.
Liu Y, Asan, Ma D, Lv F, Xu X, Wang J, Xia W, Jiang Y, Wang O, Xing X, Yu W, Wang J, Sun J, Song L, Zhu Y, Yang H, Wang J, Li M. Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int. 2017;28:2985–95.
Umair M, Alhaddad B, Rafique A, Jan A, Haack TB, Graf E, Ullah A, Ahmad F, Strom TM, Meitinger T, Ahmad W. Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr Res. 2017;82:753–8.
Won JY, Jang WY, Lee HR, Park SY, Kim WY, Park JH, Kim Y, Cho TJ. Novel missense loss-of-function mutations of WNT1 in an autosomal recessive osteogenesis imperfecta patient. Eur J Med Genet. 2017;60:411–5.
Caparros-Martin JA, Aglan MS, Temtamy S, Otaify GA, Valencia M, Nevado J, Vallespin E, Del Pozo A, Prior de Castro C, Calatrava-Ferreras L, Gutierrez P, Bueno AM, Sagastizabal B, Guillen-Navarro E, Ballesta-Martinez M, Gonzalez V, Basaran SY, Buyukoglan R, Sarikepe B, Espinoza-Valdez C, Cammarata-Scalisi F, Martinez-Glez V, Heath KE, Lapunzina P, Ruiz-Perez VL. Molecular spectrum and differential diagnosis in patients referred with sporadic or autosomal recessive osteogenesis imperfecta. Mol Genet Genomic Med. 2016;5:28–39.
Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P, Kornak U, Phadke SR. Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet. 2015;58:21–7.
Acknowledgements
We thank the patient and family members for participating in this study.
Funding
This study was supported by the Chulalongkorn Academic Advancement into Its second Century Project and the Thailand Research Fund for collection, analysis and interpretation of the data (BRG5980001; DPG6180001).
Availability of data and materials
All data are contained in the manuscript.
Author information
Authors and Affiliations
Contributions
CK and DK examined the patients, collected the samples, and drafted the manuscript. VS and KS designed the study and revised the manuscript. CS and AS analyzed the data. All authors revised and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was obtained from the institutional review board, Faculty of Medicine, Chulalongkorn University. After explanation of the possible consequences of this study, the written informed consent was obtained.
Consent for publication
The parents gave permission for the publication of the patient’s clinical details and images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kuptanon, C., Srichomthong, C., Sangsin, A. et al. The most 5′ truncating homozygous mutation of WNT1 in siblings with osteogenesis imperfecta with a variable degree of brain anomalies: a case report. BMC Med Genet 19, 117 (2018). https://doi.org/10.1186/s12881-018-0639-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0639-0