
Abstract
Background
Birt-Hogg-Dubé syndrome is an autosomal dominant hereditary condition caused by mutations in the folliculin-encoding gene FLCN (NM_144997). It is associated with skin lesions such as fibrofolliculoma, acrochordon and trichodiscoma; pulmonary lesions including spontaneous pneumothorax and pulmonary cysts and renal cancer.
Methods
Genomic DNA was extracted from peripheral venous blood samples of the propositi and their family members. Genetic analysis was performed by whole exome sequencing and Sanger sequencing aiming at corresponding exons in FLCN gene to explore the genetic mutations of these two families.
Results
In this study, we performed genetic analysis by whole exome sequencing and Sanger sequencing aiming at corresponding exons in FLCN gene to explore the genetic mutations in two Chinese families. Patients from family 1 mostly suffered from pneumothorax and pulmonary cysts, several of whom also mentioned skin lesions or kidney lesions. While in family 2, only thoracic lesions were found in the patients, without any other clinical manifestations. Two FLCN mutations have been identified: One is an insertion mutation (c.1579_1580insA/p.R527Xfs on exon 14) previously reported in three Asian families (one mainland family and two Taiwanese families); while the other is a firstly reviewed mutation in Asian population (c.649C > T / p.Gln217X on exon 7) that ever been detected in a French family.
Conclusions
Overall, The detection of these two mutations expands the spectrum of FLCN mutations and will provide insight into genetic diagnosis and counseling of Birt-Hogg-Dubé syndrome.
Similar content being viewed by others

Background
Birt-Hogg-Dubé syndrome (BHDS) is an autosomal dominant hereditary condition associated with skin lesions such as fibrofolliculoma, acrochordon and trichodiscoma, pulmonary lesions including spontaneous pneumothorax and pulmonary cysts and renal cancer. In 1925, Burnier and Rejsek reported an elderly female with multiple small skincolored papules on the head and neck, which was probably the first case of BHD [1]. In 1960, Zackheim and Pinkus described five more cases with similar clinical manifestations and histopathologic features [2]. In 1977, Birt, Hogg, and Dubé found that a few members of a thyroid cancer family had fibrofolliculoma that occurred in an autosomal dominant hereditary pattern [3]. In 2001, the susceptible locus was localised to chromosome 17p11.2 [4, 5]. Subsequently, protein-truncating mutations were identified in the FLCN (BHD) gene comprising 14 exons and encoding a protein called folliculin with unknown function [6]. Folliculin is expressed in most tissues including the skin and its appendages, the lungs (type 1 pneumocytes) and the kidney (distal nephron). Although the accurate function of this protein has not yet been clarified, it seems to be involved in the adenosine-monophosphate-activated protein kinase and mTOR pathways [7, 8]. Some studies have proved that downstream molecules of insufficient FLCN such as S6 kinase and hypoxia-inducible factor 1-alpha (HIF-1a) increases in renal tumors derived from BHDS patients. In the lung, cyst-lining cells were suggested to be activated due to their immunostaining positivity for phospho-mTOR and phospho-S6 ribosomal protein [9,10,11,12]. As neoplastic hyperplasia hardly occurs in cyst-lining cells, the mTOR pathway may be less distinctively detected in pulmonary cysts [11].
More than 200 mutations in the FLCN gene have been identifed, most of which are frameshift, nonsense, missense, or splice site mutations. The most common mutation in patients with Birt-Hogg-Dubé syndrome is c.1285dupC located in exon 11 [13,14,15,16,17,18,19,20,21,22], followed by c.1533_1536delGATG [12, 15, 23,24,25] and c.1278dupC [26,27,28,29] depending on literatures listed worldwide up to date. Table 1 presents the mutations described in the FLCN gene up to now according to literatures summarized by searching “Birt-Hogg-Dubé syndrome” and “FLCN” on pubmed and Embase line.
Objectives
The aim of this study is to explore the genetic mutations of two suspected BHDS families, and to see if they could expand the spectrum of FLCN mutations.
Methods
The two BHDS families were recruited from Peking Union Medical College Hospital and Xiangya Hospital Central South University. Detailed physical examination and other relevant examination of the participants were carried out. Peripheral venous blood samples of the participants were collected with anticoagulant tubes, storage and transportation of which were under the condition of 4 °C, then genomic DNA was extracted from blood samples within 6 h for further gene analysis: The whole blood and erythrocyte lysate were mixed thoroughly, kept still on ice for about 30 min until clear and then centrifuged at 3000 rpm for 10 min (4 °C); abandoned the supernatant, and mixed the remnant with nuclear lysate. Then added proteinase K into the mixture and mixed them thoroughly until there was no cell precipitate. Added SDS and shook at 37°Cfor 6 h or overnight. Added saturated phenol, mixed well up and down and centrifuged at 3000 rpm for 10 min (4 °C). Then put the supernatant into the mixture of saturated phenol and chloroform (1: 1), mixed well up and down and centrifuged at 3000 rpm for 10 min (4 °C); after that, put the supernatant into chloroform, mixed thoroughly up and down and centrifuged at 3000 rpm for 10 min (4 °C). The supernatant was added to a centrifuge tube previously charged with ethanol, gently inverted it to precipitate the DNA. The DNA and a small amount of ethanol was transferred to an eppendorf tube finally and stored at −20 °C in reserve.
With clinical manifestations and family history of pneumothorax, the patients and some of their relatives were diagnosed with suspected BHDS, at the meantime, unaffected relatives were invited to participate as controls. Members II10, III8, III10, III11, III12, III13, III14, IV1, IV2, IV3, IV4 in family 1 and II1, III2 in family 2 were sequenced. Publication of all the medical data has obtained consent of the participants, and the propositi consented on behalf of the deceased patients to both participate and to have their data published.
We selected one patient from each family respectively (IV3 in family 1 and III2 in family 2), carrying out whole exome sequencing for mutation detection: The 300 ng genomic DNA concentrations were sheared with Covaris LE220 Sonicator (Covaris) to target of 150-200 bp average size. DNA libraries were prepared using SureselectXT reagent kit (Agilent). The fragments were repaired the 3′ and 5′ overhangs using End repair mix (component of SureselectXT) and purified using Agencourt AMPure XP Beads (Beckman). The purified fragments were added with’A’ tail using A tailing Mix (component of SureSelectXT) and then ligated with adapter using the DNA ligase (component of SureselectXT). The adapter-ligated DNA fragments were amplified with Herculase II Fusion DNA Polymerase (Agilent). Finally, the pre-capture libraries containing exome sequences were captured using SureSelect capture library kit (Agilent). DNA concentration of the enriched sequencing libraries was measured with the Qubit 2.0 fluorometer dsDNA HS Assay (Thermo Fisher Scientific). Size distribution of the resulting sequencing libraries was analyzed using Agilent BioAnalyzer 2100 (Agilent). The libraries were used in cluster formation on an Illumina cBOT cluster generation system with HiSeq PE Cluster Kits (illumina). Paired-end sequencing is performed using an Illumina HiSeq system following Illumina-provided protocols for 2 × 150 paired-end sequencing. Then we applied Sanger sequencing aiming at corresponding exons in FLCN gene for subsequent validation of other family members roughly as follows: PCR amplification with appropriate primers on PCR amplifier - PCR cleanup in magnetic bead purification system - cycle sequencing on PCR amplifier - sequencing cleanup on magnetic bead purification platform - capillary electrophoresis on ABI3730. Interpretation of Sanger sequencing results was performed using SnapGene Software.
Results
Family 1 (F1)
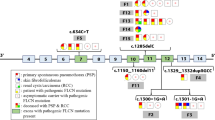
The proband, a 47-year-old woman with a 25-year history of left-lung-pneumothorax, has had her left lung partially resected. Moreover, she was diagnosed with cerebral infarction 3 years ago on account of right limb numbness and visual defect in the lower half of the right eye. In addition, two of her sisters and their sons (Fig. 1: III8, III12, IV1, IV3) also had spontaneous pneumothorax history at the age of 39, 48, 21 and 21 respectively, a maximum frequency of which was six times. Diffuse lesions of the thyroid gland, superficial lymph node enlargement of the neck and extremities and subcutaneous nodules of the head, neck and hands were also revealed in one of her sister (III8) after pulmonary bubble resection; computed tomography (CT) scans of the other sister (III12) who had a history of hysteromyoma excision ever showed double renal cysts, which disappeared 2 years later in the renal ultrasonic examination results. While one nephew (IV1) of the proband had fat granules on his face and neck, who once underwent right branchial cystectomy; the other nephew (IV3) was diagnosed with chronic pancreatitis at 11 years old. A few of her other family members (Fig. 1:II1, II4, II7, II9, II11; II9: cerebral hemorrhage, others: cerebral infarction) also suffered from stroke, all of whom have passed away. One died of thrombocythemia (Fig. 1:III1). (Fig. 2).

Pedigre of family 1.  proband.
proband.  Cases with stroke.
Cases with stroke.  Thrombocythemia Case
Thrombocythemia Case

Examination results and Sequence diagram of family 1. a, b, c Computed tomography scans showing multiple cystic lesions in the lungs of patients (III8、III12、III14). b, e Computed tomography scan and X-ray examination results showing pneumothorax (III8、IV3). d Fat granules on the skin (IV1). f Direct sequencing of exon 14 of FLCN revealed the frameshift mutation: c.1579_1580insA on exon 14
Family 2 (F2)
A 26-year-old man with after-exercise pectoralgia was diagnosed pneumothorax with CT scans, and before that, he once had a pneumothorax attack. In his family members, his father and grandfather also had pneumothorax history, for which his father had a thoracoscopic surgery. Besides, his grandfather passed away because of nephropathy without concrete information (Figs. 3 and 4). The clinical information of the two families are listed in Table 2.

Pedigre of family 2.  proband
proband

Examination results and Sequence diagram of family 2. a Computed tomography scans showing pulmonary cyst and pneumothorax (III2). b, c Multiple pulmonary cysts and pneumothorax in the lung of the proband’s father (II1). d, e Direct sequencing of exon7 of FLCN revealed the nonsense mutation: c.649C > T on exon 7
Mutation examinations revealed that the proband, her two sisters, two nephews (III8, III12, IV1, IV3) and her son (IV4) in F1 all carried a one-base (A) -insertion between nucleotides c.1579_1580 on exon 14 (c.1579_1580insA) (Fig. 2f), resulting in a frameshift mutation (p.Arg527Xfs), which has ever been reported in three Asian families [30,31,32]; while the proband and his father in F2 carried a one-base-substitution of C by T at nucleotide c.649 on exon 7 (c.649C > T) (Fig. 4d, e), resulting in a nonsense mutation (p.Gln217X), which was once recovered in a French family [22]. In addition, there are no mutations detected in the control subjects (II10, III10, III11, III13, IV2).
Discussion
Studies of patients with Birt-Hogg-Dubé syndrome are very rare especially in Asian countries.
In this study, we described two BHDS families and applied whole exome sequencing and Sanger sequencing to explore the genetic mutations. Patients from family 1 mostly suffered from pneumothorax and pulmonary cysts, several of whom also mentioned skin lesions or kidney lesions. While in family 2, only thoracic lesions were found in the patients, without any other clinical manifestations. Two FLCN mutations have been identified: One is an insertion mutation (c.1579_1580insA/p.R527Xfs) previously reported in three Asian families (one mainland family and two Taiwanese families); while the other is a firstly reviewed mutation in Asian population (c.649C > T/p.Gln217X) that ever been detected in a french family.
As we have reported above, patients from these two families were mostly characterized by pneumothorax, and even without any other clinical manifestations, which may remind us of BHDS and carrying out genetic tests for patients with familial pneumothorax history. However, the exact mechanism of this syndrome is still unclear till now. Our study could only expand the spectrum of FLCN mutations ethnically, there are still many aspects of BHDS to be explored.
Conclusions
Our detection of these two mutations expands the spectrum of FLCN mutations and will provide insight into genetic diagnosis and counseling of Birt-Hogg-Dubé syndrome.
Abbreviations
- BHDS:
-
Birt-Hogg-Dubé syndrome
References
Rejsek B. Fibromes sous-cutanes peripilares multiples du cou. Bull Soc Fr Dermatol Syphiligr. 1925;32:242–3.
Zackheim HS, Pinkus H. Perifollicular Fibromas. Arch Dermatol. 1960;82:913–9.
Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113(12):1674e7.
Khoo SK, Bradley M, Wong FK, Hedblad M-A, Nordenskjöld M, Teh BT. Birt-Hogg-Dubé syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. 2001;20:5239–42.
Schmidt LS, Warren MB, Nickerson ML, et al. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–82.
Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157–64.
Hartman TR, Nicolas E, Klein-Szanto A, Al-Saleem T, Cash TP, Simon MC, et al. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28:1594–604.
Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein FNIP1 and AMPK, and is involved in AMPK and MTOR signalling. Proc Natl Acad Sci U S A. 2006;103:15552–7.
Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Familial Cancer. 2013;12(3):387e96.
Ponti G, Pellacani G, Seidenari S, Pollio A, Muscatello U, Tomasi A. Cancer-associated genodermatoses: skin neoplasms as clues to hereditary tumor syndromes. Crit Rev Oncol Hematol. 2013;85(3):239e56.
Nishii T, Tanabe M, Tanaka R, Matsuzawa T, Okudela K, Nozawa A, et al. Unique mutation, accelerated mTOR signaling and angiogenesis in the pulmonary cysts of BirtHogg-Dube’syndrome. Pathol Int. 2013;63(1):45e55.
Furuya M, Tanaka R, Koga S, Yatabe Y, Gotoda H, Takagi S, et al. Pulmonary cysts of Birt-Hogg-Dubé syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am J Surg Pathol. 2012;36(4):589e600.
Liu Y, Xu Z, Feng R, et al. Clinical and genetic characteristics of chinese patients with Birt-Hogg-Dubé syndrome. Orphanet J Rare Dis. 2017;12(1):104.
Park HJ, Park CH, Lee SE, et al. Birt-Hogg-Dubé syndrome prospectively detected by review of chest computed tomography scans. PLoS One. 2017;12(2):e0170713.
Furuya M, Yao M, Tanaka R, et al. Genetic, epidemiologic and clinicopathologic studies of Japanese Asian patients with Birt-Hogg-Dubé syndrome. Clin Genet. 2016;90(5):403–12.
Yamada Y, Sakamoto S, Furuya M, et al. Case of bilateral and multifocal renal cell carcinoma associated with Birt-Hogg-Dubé syndrome. Int J Urol. 2015;22(2):230–1.
Spring P, Fellmann F, Giraud S, et al. Syndrome of Birt-Hogg-Dubé, a histopathological pitfall with similarities to tuberous sclerosis: a report of three cases. Am J Dermatopathol. 2013;35(2):241–5.
Kashiwada T, Shimizu H, Tamura K, et al. Birt-Hogg-Dubé syndrome and familial adenomatous polyposis: an association or a coincidence? Intern Med. 2012;51(13):1789–92.
Shin WW, Baek YS, Oh TS, et al. Birt-Hogg-Dubé syndrome, a rare case in Korea confirmed by genetic analysis. Ann Dermatol. 2011;23(Suppl 2):S193–6.
Houweling AC, Gijezen LM, Jonker MA, et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dubé syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011;105(12):1912–9.
Maffé A, Toschi B, Circo G, et al. Constitutional FLCN mutations in patients with suspected Birt-Hogg-Dubé syndrome ascertained for non-cutaneous manifestations. Clin Genet. 2011;79(4):345–54.
Kluger N, Giraud S, Coupier I, et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol. 2010;162(3):527–37.
Kuroda N, Furuya M, Nagashima Y, et al. Intratumoral peripheral small papillary tufts: a diagnostic clue of renal tumors associated with Birt-Hogg-Dubé syndrome. Ann Diagn Pathol. 2014;18(3):171–6.
Furuya M, Nakatani Y. Birt-Hogg-Dubé syndrome: clinicopathological features of the lung. J Clin Pathol. 2013;66(3):178–86.
Kunogi M, Kurihara M, Ikegami TS, et al. Clinical and genetic spectrum of Birt-Hogg-Dubé syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet. 2010;47(4):281–7.
Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321–31.
Ren HZ, Zhu CC, Yang C, et al. Mutation analysis of the FLCN gene in Chinese patients with sporadic and familial isolated primary spontaneous pneumothorax. Clin Genet. 2008;74(2):178–83.
Schmidt LS, Nickerson ML, Warren MB, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet. 2005;76(6):1023–33.
Kawasaki H, Sawamura D, Nakazawa H, et al. Detection of 1733insC mutations in an Asian family with Birt-Hogg-Dubé syndrome. Br J Dermatol. 2005;152(1):142–5.
Furuya M, Tanaka R, Koga S, et al. Pulmonary cysts of Birt-Hogg-Dubé syndrome: a clinicopathologic and immunohistochemical study of 9 families. Am J Surg Pathol. 2012;36(4):589–600.
Yang CY, Wang HC, Chen JS, et al. Isolated familial pneumothorax in a Taiwanese family with Birt-Hogg-Dubé syndrome. J Postgrad Med. 2013;59(4):321–3.
Liu L, Yang K, Wang X, et al. Detection of Folliculin gene mutations in two Chinese families with Birt-Hogg-Dubé syndrome. Biomed Res Int. 2017;2017:8751384.
Funding
This study was supported by the National Key Research and Development Program of China (Nos. 2016YFC0901504, 2016YFC0905100, to Hong Jiang), the National Natural Science Foundation of China (Nos. 81471156, 81771231, to Hong Jiang), the National Natural Science Foundation of China (No.31401135, to Rong Qiu), High-level medical personnel of Hunan province “225” Project and Clinical Research Funds of Xiangya Hospital (No. 2014 L03, to Hong Jiang), the Natural Science Foundation of Hunan Province, China (No. 2017JJ2345, to Wei Zhuang). We are grateful for the assistance from Wuxi AppTec and TsingKe Biological Technology service.
Availability of data and materials
The data and materials generated during the study are available from the corresponding author on reasonable request. The datasets generated during the current study are available at the Sequence Read Archive (SRA) repository under accession code SRP127011. Confidential patient data has not been shared.
Author information
Authors and Affiliations
Contributions
Guarantor of integrity of entire study: WZ, HJ, KX, BS T; Study design: WZ and HJ; Literature research: XC H; Clinical studies: YZ; Experimental studies: XC H and YP; Data acquisition: YZ and YP; Data analysis/interpretation: XC H; Statistical analysis: XC H and YZ; Manuscript preparation: XC H and HJ; Manuscript defnition of intellectual content: WZ and YZ; Manuscript editing: XC H; Manuscript revision/review: RQ, KX; BS T; Experimental condition and facilities provision: KX; BS T; Final approval of the version to be published: all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This research has been approved by Medical Ethics Committee of Xiangya Hospital Central South University, China with the reference number of 201709983 (IRB(s) No.). Informed consent of all participants has been obtained, and the propositi consented on behalf of the deceased patients to both participate and to have their data published.
Consent for publication
Publication of all the medical data included in this article has obtained consent of the participants, and the propositi consented on behalf of the deceased patients to both participate and to have their data published.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hou, X., Zhou, Y., Peng, Y. et al. Birt-Hogg-Dubé syndrome in two Chinese families with mutations in the FLCN gene. BMC Med Genet 19, 14 (2018). https://doi.org/10.1186/s12881-017-0519-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0519-z