
Abstract
Background
SCN8A mutations have recently been associated with epilepsy and neurodevelopmental disorders. This study aimed to broaden the phenotypic-spectrum of disease related with SCN8A mutations.
Methods
To identify the pathogenic gene of a Chinese family, in which six members suffered from epilepsy, whole-exome sequencing was performed. In addition, target next-generation sequencing (NGS) was performed on 178 sporadic patients, who had epilepsy of unknown etiology within 6 months after birth. A detailed clinical history was obtained.
Results
A heterozygous missense mutation of SCN8A was identified in the Chinese family. Six de novo mutations of SCN8A were detected in 6 sporadic patients with epilepsy. In the family, six members developed seizures within a few years after birth. Five of them had milder clinical performance, that they had normal cognition and developmental milestones, and seizure-free was achieved by mono-therapy. The other one affected member presented with refractory epilepsy and developmental regression. She died from sudden unexpected death in epilepsy (SUDEP) at 17-year-old. Clinical features of six sporadic patients with SCN8A mutations were diverse, ranging from severe epileptic encephalopathy to benign epilepsy with normal cognition. Seizures started at the mean age of 3.9 months (from 2 months to 6 months). Seizure-free was achieved in four of them by mono- or multi-antiepileptic drugs. Five of them demonstrated mild or severe psychomotor retardation, whereas the other one was normal in development and intelligence.
Conclusions
Our findings extend the spectrum of SCN8A mutations and the clinical features of patients with SCN8A mutations. The majority of SCN8A mutations were de novo, inherited mutations from the heterozygous parents can also occur. The phenotypic spectrum of SCN8A mutation varied largely. Most affected patients manifested as refractory epilepsy and severe intellectual disability, only a small number of patients presented with milder clinical patterns. Additionally, our study confirmed that the same mutation can lead to different phenotypes.
Similar content being viewed by others
Background
Voltage-gated sodium channels (VGSCs) play a critical role in controlling neuronal excitability. VGSCs contain one pore-forming α-subunit and one or two β subunits. The α-subunit is made up of four repeated homologous domains (I ~ IV), each domain containing a motif of six transmembrane segments (S1 ~ S6) and an additional pore loop located between the S5 and S6 segments [1]. So far, 9 VGSCs α-subunits (Nav1.1 ~ Nav1.9) have been identified. Among these 9 α-subunits, Nav1.1, 1.2, 1.3 and 1.6, which are encoded by SCN1A, SCN2A, SCN3A and SCN8A respectively, are primarily expressed in the central nervous system. Mutations in these genes can result in epilepsy [2,3,4,5].
The Nav1.6 α-subunit is encoded by SCN8A, which is highly expressed along the axonal initial segment (AIS) and the nodes of Ranvier of myelinated axons [6, 7]. Nav1.6 is directly involved in action potential generation and conduction. Mutations of SCN8A are associated with type 13 of early infantile epileptic encephalopathy (EIEE 13) (OMIM #614558) [8, 9]. The phenotypic spectrum of diseases caused by SCN8A mutations varied largely. Most patients had intractable epilepsy beginning at the first year of life, accompanied by severe developmental delay and intellectual disability. SCN8A mutations can also lead to milder phenotype, such as benign infantile seizures (BFIS)/infantile convulsion and paroxysmal choreoathetosis (ICCA) [10]. Part of patients with SCN8A mutations presented only mental retardation without epilepsy [11]. In this study, seven SCN8A mutations were identified in a Chinese family and six sporadic patients with unknown etiological epilepsy, including four previously described mutations and three novel mutations.
Methods
Patients and samples
A Chinese family was recruited in this study, in which six members spanning 3 generations were affected with epilepsy (Fig. 1). All of them were female, and aged from 2 to 61 years old. PCDH19 gene mutation was ruled out in this family. The proband had focal seizures at the age of 6 months. Seizure frequency ranged from one time per month to 6 times per day. She was treated with valproic acid (VPA) at 8 months of age. Seizure-free was achieved at 1 year old. III-5 had similar clinical course as the proband. Seizures attacked at 6-month-old, which was also controlled by VPA.II-2 and I-2 suffered from febrile convulsions during childhood, which evolved into afebrile seizures after 10 years old for both of them. Seizure-free was achieved in both of them, by treatment with Phenytoin (PHT) and Carbamazepine (CBZ) respectively. II-4 had focal seizures during the preschool period, and there were no seizures reported since six years old, even without treatment. Another member (II-3) in this family had been suffering from epilepsy since she was less than one-year-old. Unfortunately, seizures were not controlled by CBZ. Early development was normal for her. However, after seizures onset, she had significant retrogression in cognition, and gradually lost the ability of walking and speaking. At 17 years old, she died from sudden unexplained death in epilepsy (SUDEP). Except II-3, other five affected members in the family had normal cognition and motor function, and neurological examinations were unremarkable. Detailed clinical information of the affected individuals in this family was summarized in Table 1 and Additional file 1.

The pedigree of family case with SCN8A mutation.I-2, II-2, II-4, III-2, III-4, were analyzed by whole-exome sequencing. III-5 was tested by Sanger sequencing
In addition, a total of 178 sporadic patients having unknown etiological epilepsy were recruited. All of the patients had epilepsy, and met one or more of the following criteria: 1) seizures onset within 6 months, especially within 3 months after birth; 2) intractable epilepsy with frequent seizures; 3) severe electroencephalography (EEG) abnormalities, including hypsarrhythmia, burst-supperession, etc.; 4) severe psychomotor retardation, with or without regression. We excluded the patients with neonatal or infant epileptic seizures caused by metabolic diseases, brain injury during perinatal period, central nervous system infection or other known etiologies.
The detailed clinical information was collected by periodic follow-up, including medical history, family history, curative effects by antiepileptic drugs (AEDs), MRI, EEG, etc.,. Genomic DNA was extracted from peripheral leukocytes. Written informed consent was obtained from the subjects. This study was approved by the Peking University First Hospital Medical Ethics Committee.
Whole-exome sequencing
Whole-exome sequencing was used to detect the mutated gene in the family. Probes were designed to cover the whole-exome. Sequencing was performed on Ion torrent Proton high-throughput platform (Themofisher) using a paired-end sequencing of 200 bp to screen the mutations. Fast QC v0.10.1 was used to filter out reads of low base quality score. BWA software was used to align the reads to the hg19 genome. Unified Genotyper and HapolotypeCaller in Genome Analysis Tool KIT v3.2 (GATKv3.2) were used to call variants.
Targeted NGS
Targeted NGS was used to screen gene mutations in 178 sporadic patients with epilepsy of unknown cause. A custom-designed panel capturing the coding exons of 485 genes (Additional file 2) related to epilepsy and intellectual disability, including SCN8A, was synthesized using NimbleGenSeqCap Target Enrichment technique. NGS was subsequently performed on Ion torrent Proton high-throughput platform (Themofisher).
Mutations annotation
All the mutations were confirmed by Sanger sequencing. Rare mutations, whose population frequency was less than 1%, were filtered according to the 1000 Genomes data, ESP6500 population data and ExAC data. Reported pathogenic mutations in HGMD Professional database and Pubmed were marked. The pathogenicity of other rare mutations was annotated by Mutation Taster and PolyPhen-2.
Sanger sequencing was performed to analyze the parental origin of those variants. Then the causative mutations were confirmed according to parental origin of the variants and the clinical features of the patients.
Results
Molecular analysis
Five members, including four patients (I-2, II-2, II-4, III-4) and one phenotypic normal member (III-2) in the Chinese family were analyzed by whole-exome sequencing (Fig. 1). Meanwhile, patient III-5 was not born yet. A total of 23 candidate gene mutations were identified (Additional file 3). After III-5 was born and presented with seizures at age of 6 months, Sanger sequencing of these 23 candidate genes was performed on the newborn. Then it was narrowed to 10 candidate genes including SCN8A and 9 other genes (Table 2). Among these candidate genes, 7 genes were neither expressed in nervous system, nor reported to be associated with epilepsy or other neurologic disorders. ANGPT4 has been related to Alzheimer’s disease and vascular dementia. IMPA1 has been associated with bipolar disorder previously. But the variants of them detected in this family were also frequent in ExAC and 1000 Genomes database. SCN8A has been related with EIEE 13, benign infantile epilepsy, and cerebellar ataxia. It is widely expressed in central nervous system and had been reported in patients with epilepsy. The heterozygous missense mutation (c.4793 T > C, p.Val1598Ala) of SCN8A identified in this family was not mentioned in any known database (dbSNP138, 1000G, EVS, ExAC). The mutation site was highly conserved from insects to mammals (Fig. 2h), and was predicted as damaging by Mutation Taster and PolyPhen-2 (Table 3). So SCN8A was considered to be the causative gene of the family.

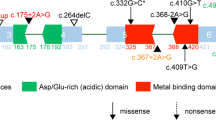
Seven SCN8A mutations identified in this study a-g and conservation of the altered amino acid of three unreported mutations shown in the ClustalW alignments h
By targeted NGS, heterozygous mutations in seven genes (CDKL5, KCNQ2, KCNT1, STXBP1, SCN2A, SCN8A, SLC2A1) were detected in 59 patients. The gene mutation rate was 33% (59/178). SCN8A gene mutations were identified in six patients (Fig. 2a-f). All six mutations were de novo, which were predicted highly damaging the function of Nav1.6 channel by Mutation Taster and PolyPhen-2 (Table 2). Four of them have been confirmed pathogenic previously (Table 3). The other two novel SCN8A mutations (c.694 T > C, p.Ser232Pro and c.2671G > A, p.Val891Met) are located at the extremely conserved positions (Fig. 2h). A schematic representation of the Nav1.6 channel, including the locations of mutations identified in this study, is shown in Fig. 3.

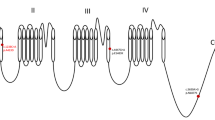
The structure of the human Nav1.6 channel and the location of SCN8A mutations identified in this study. Red Dots: Novel; black dots: reported
Clinical and neurophysiological findings
Family case
The clinical information of six affected patients was summarized in Table 1.
Sporadic cases
Six patients were identified having SCN8A mutations, including four males and two females. The clinical features of them were diverse, of whom four presented severe epileptic encephalopathy, whereas the other two had much milder clinical course. The age of epilepsy onset ranged from 2 month to 5 month, averaging at 3.9 months. Focal seizure was the most common semiology, observed in all six of them. Seizure-free was achieved in four of them eventually.
Patient 1 had focal seizures beginning at two months after birth. Seizures attacked 2–3 times every day. He was treated with Topiramate (TPM) at 3 months of age, and achieved a seizure-free period as long as 45 days. At 5 months old, seizures recurred, and the frequency was 10 times per day. Levetiracetam (LEV) was used but without remission. Then VPA was added, and seizure frequency was reduced to 5–6 times per day. TPM was stopped at 8 months old, and Clonazepam (CZP), Oxcarbazepine (OXC) was added at 11 months of age. By treatment with multi-drugs (VPA, OXC, CZP), no seizures were observed since from one year of age. He presented with severe developmental delay and is currently (32 months of age) unable to sit independently.
Patient 2 was discovered to have epilepsy at 3 months of age. The initial seizure type was focal seizure. Seizures attacked 7–8 times per day. He was treated with LEV at 5 months, but no good outcome was reported. At 9 months of age, myoclonic seizures were observed. EEG at that time showed generalized high amplitude multi-spike and slow waves. At age of 1 year and 7 months, LEV was substituted by VPA, and seizure frequency was reduced to 3 times per day. By 1 year and 11 months, Lamotrigine (LTG) was added for treatment, and no seizures were reported after 2 years old. The developmental milestones were far behind his peers, that he could not control his head until 28 months old.
Patient 3 began to have seizures at age of 3.5 months, including focal seizures, secondary generalized seizures, and status epilepticus. It was as frequent as 10–15 times per day. EEG at 1-year-old showed slow background and spikes at right central area during sleep. He remained have seizures at age of one year, even with multi-therapy (LEV, TPM, VPA and OXC). Developmental delays caused concern since from 3 months of age, when poor head control was observed, and there was no apparent improvement throughout the follow-up period (one-year-old). As patient 3 was loss to follow-up, his current status was not clear.
Patient 4 developed epilepsy at 4 months old. The initial seizure type was focal seizures. Generalized seizures were also observed. Seizures attacked 1–2 times per week. She was treated with VPA at 5 months of age, but showed no signs of remission. Then TPM was added at 6 months old. By treatment with VPA and TPM, she had no seizures for 6 months (from 6-month-old to 1-year-old). However, seizures attacked again at 1 year old and the frequency was as high as 1–2 times per day. By that time, CZP was added but with no effects. CBZ was added at 2 years and 2 months old, by which seizure frequency was markedly reduced, and the intermittent phase could be as long as 50 days. EEG was at boundary condition at 2 years of age. She presented with severe hypotonia and couldn’t control head at age of 26 months. After the seizure frequency was reduced, she showed significant developmental progress, that she could control head at 2 years and 5 months.
Patient 5 demonstrated milder phenotype. Epilepsy onset was at 5 months old, with focal seizures followed by secondary generalization. EEG at age of 8 months showed sharp and spike waves in the right frontal during sleeping period, and 3–4 Hz slow-wave complexes in the occipital region during awake period. Seizures attacked 1–2 times per day. VPA and LEV were not effective at age of 8 months. Then OXC was added at that time, no seizure was reported from then on. Developmental milestones were normal before the onset of seizures, with head control at 2 months of age. After seizures onset, her development was slightly delayed that she could not sit independently until 9 months. Patient 5 didn’t show obvious cognition impairment.
Patient 6 had focal seizures starting at 6 months old. Seizures attacked once every month. At 11 months old, he was treated with LEV, but seizures still occurred occasionally. OXC was used at 18 months of age, and since then no seizures were observed. Then two months later LEV was stopped. He presented with normal development and cognition.
The detailed clinical information of the six sporadic cases was summarized in Table 4 and Additional file 1.
Discussion
A total of seven SCN8A mutations were identified, of which three were novel. These new patients provide support for delineating the clinical features of patients with SCN8A mutations.
SCN8A mutations have already been proved responsible for EIEE 13. Generally, patients had seizure onset within 6 months after birth [9]. The seizure semiology was variable, including focal seizures, clonic seizures, tonic-clonic seizures, epileptic spasms, myoclonic and absence seizures, etc. [9, 12]. The majority of affected patients had mild to severe psychomotor retardation. Hypotonia, hypertonia and dystonia were common as well [9]. Patients with SCN8A mutations also have a high incidence of SUDEP [13, 14]. In our study, the clinical profiles of four patients (Patient 1–4) were in accordance with the characteristic of EIEE13. Seizures began at a mean age of 3.5 months and were intractable. Focal seizure was the primary seizure type. All of them suffered from severe developmental delay and intellectual disability (Table 4). Hypotonia was also obvious. The other two patients (patient 5, and 6) presented with milder phenotype, which confirmed that diseases caused by SCN8A mutations had a broad phenotypic-spectrum.
Four mutations (those of patient 2, 3, 4, and 5) had been described previously. The clinical features of patient 2, 3, and 4 were quite similar as the characterization of patients with each same mutation previously described [12, 14, 15]. The phenotype of patient 5 was much milder. With multi-drugs (LEV, VPA, OXC), no seizures were observed since 8 months old. In addition, she didn’t present obvious cognition impairment, whereas her phenotype was slightly more serious than the patients with the same mutation (c.5630A > G, p. Asn1877Ser) in Anand’s report [16]. Anand et al. reported a family with SCN8A mutation, in which the proband and his father had early onset focal epileptic seizures without cognitive or neurological impairment. The seizures were controlled well by mono-therapy, with CZB and PHT, respectively. Additionally, in the report of Parrini et al., patients with the same mutation (c.5630A > G, p. Asn1877Ser) presented with drug resistant focal epilepsy and mild intellectual disability [15]. Nevertheless, this mutation has also been reported to cause more severe phenotype including intractable epilepsy, developmental delay and intellectual disability [16]. This study and previous reports suggest that same mutation in SCN8A can lead to a different phenotype.
Patient 6, with mutation (c.2671G > A, p.Val891Met) in SCN8A, had a benign clinical course. He presented no psychomotor retardation, and epilepsy was well controlled by mono-therapy. It was the first reported de novo SCN8A mutation relating with benign phenotype. Combined with previous reports, our study provided further evidence that SCN8A mutations could lead to a wild range of phenotypes.
To date, more than 100 SCN8A mutations have been reported, an overwhelming majority of which were de novo [12, 14, 17,18,19,20,21,22,23,24,25,26,27,28,29]. Only few inherited SCN8A mutations were reported. Trudeau et al. first reported a frame-shift mutation (c.5156_5157delCT, p.Pro1719fs) of SCN8A in a family with mental retardation and ataxia, but without epilepsy [11]. Gardella et al. discovered a SCN8A mutation (c.4447G > A, p.Glu1483Lys) in 16 affected members of three families with BFIS/ICCA [10]. Anand et al. identified a heterozygous mutation (c.5630A > G, p.Asn1877Ser) of SCN8A in a family with early onset epilepsy with no cognitive impairment [16]. The heterozygous mutation (c.4793 T > C, p.Val1598Ala) identified in our Chinese family was the forth reported inherited SCN8A mutation. In addition, all patients in this family were female, and PCDH19 gene mutation was ruled out. The onset age ranged from 6 months to preschool period. Two (I-2 and II-2) of them initialized as febrile convulsion, which evolved into afebrile seizures. The others initialized as focal seizures. Good seizure control was achieved in four of them with mono-therapy, and II-4 was seizure free even without treatment. While seizures were refractory inII-3. At the age of 17 years, she died from SUDEP. Except II-3, the other five affected members had normal cognition and developmental profile. To the best of our knowledge, this is the only family among which different phenotypes presented in the members with the same SCN8A mutation. It is proposed that other genetic or non-genetic factors account for the different phenotypes. Unfortunately, as II-3 had passed away, the difference of genetic background between the affected members, which might explain their significant variation on phenotypes, was not available.
The reported mutations are scattered throughout the whole SCN8A channel [9]. Among these mutations, Arg1872 and Arg1617 might be two mutational hotspots, which had been identified in more than ten independent patients [14, 17, 23, 26]. In vitro functional analysis proved that both of the mutations Arg1872 and Arg1617 impaired the Nav1.6 channel transition from open state to inactivated state, resulting in channel hyperactivity [30]. Like Arg1617, the mutations c.2549G > A, p.Arg850Glu and c.694 T > C, p.Ser232Pro identified in this study were located in S4 transmembrane segments (Figure 3), which served as the voltage sensors for Nav1.6 activation. So, these two mutations might have the similar pathogenic mechanism as mutations at Arg1617. They might lead to elevating Nav1.6 channel activity by impairing channel inactivation.
Sodium channel blockers may be the rational candidate drugs for the treatment of epilepsy caused by SCN8A mutations, including OXC,CBZ, LTG, Phenobarbital (PB), TPM and PHT, etc. [31, 32]. For benign familial infantile epilepsy caused by SCN8A mutation, seizures were easily controlled by mono-therapy. While for the epileptic encephalopathy, seizures were not easy to control. Approximately half of patients showed good responses to sodium channel blockers, either as a reduction in seizures frequency or even seizure-free [31, 32]. Up to 1/3 of patients were seizure-free after treatment with antiepileptic drugs [9]. In our study, four sporadic patients were seizure-free by treatment with mono- or multi- antiepileptic drugs. All of them were treated with sodium channel blockers.
Conclusions
SCN8A mutation is not only associated with epileptic encephalopathy, but also can be the pathogenic cause of some benign phenotypes, such as BFIS/ICCA, especially the inherited mutations. Three novel SCN8A mutations were identified in this study. The huge difference among the family affected members, and the varied phenotype between patient 5 and patients previously reported with same mutation (c.5630A > G, p.Asn1877Ser), suggested that the same mutation in SCN8A can lead to quite different phenotypes. It is necessary to analyze SCN8A mutations in patients with early onset epilepsy with or without developmental delay or intellectual disability. The limited clinical evidence suggests that sodium channel blockers may be the good choice for patients with SCN8A mutations.
Abbreviations
- AEDs:
-
Antiepileptic drugs
- AIS:
-
Axonal initial segment
- BFIS:
-
Benign infantile seizures
- CBZ:
-
Carbamazepine
- CZP:
-
Clonazepam
- EEG:
-
Electroencephalography
- EIEE:
-
Early infantile epileptic encephalopathy
- ICCA:
-
Infantile convulsion and paroxysmal choreoathetosis
- LEV:
-
Levetiracetam
- LTG:
-
Lamotrigine
- OXC:
-
Oxcarbazepine
- PB:
-
Phenobarbital
- PHT:
-
Phenytoin
- SUDEP:
-
Sudden unexpected death in epilepsy
- TPM:
-
Topiramate
- VGSCs:
-
Voltage-gated sodium channels
- VPA:
-
Valproic acid
References
Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25.
Scheffer IE, Zhang YH, Jansen FE, Dibbens L. Dravet syndrome or genetic (generalized) epilepsy with febrile seizures plus? Brain and Development. 2009;31(5):394–400.
Shi X, Yasumoto S, Kurahashi H, Nakagawa E, Fukasawa T, Uchiya S, Hirose S. Clinical spectrum of SCN2A mutations. Brain and Development. 2012;34(7):541–5.
Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81(11):992–8.
Vanoye CG, Gurnett CA, Holland KD, George AJ, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiol Dis. 2014;62:313–22.
Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12(8):996–1002.
Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC, Novakovic SD. Differential distribution of the tetrodotoxin-sensitive Rpn4/NaCh6/Scn8a sodium channel in the nervous system. J Neurosci Res. 2000;60(1):37–44.
Meisler MH, Helman G, Hammer MF, Fureman BE, Gaillard WD, Goldin AL, Hirose S, Ishii A, Kroner BL, Lossin C, et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia. 2016;57(7):1027–35.
Wagnon JL, Meisler MH. Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Front Neurol. 2015;6:104.
Gardella E, Becker F, Moller RS, Schubert J, Lemke JR, Larsen LH, Eiberg H, Nothnagel M, Thiele H, Altmuller J, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. 2016;79(3):428–36.
Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Genet. 2006;43(6):527–30.
Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, Depienne C, Brilstra E, Mang Y, Nielsen JE, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84(5):480–9.
Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90(3):502–10.
Kong W, Zhang Y, Gao Y, Liu X, Gao K, Xie H, Wang J, Wu Y, Zhang Y, Wu X, et al. SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia. 2015;56(3):431–8.
Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, Chiti L, Rutigliano D, Bianchini C, Virdo S, et al. Diagnostic Targeted Resequencing in 349 Patients with Drug-Resistant Pediatric Epilepsies Identifies Causative Mutations in 30 Different Genes. Hum Mutat. 2017;38(2):216–25.
Anand G, Collett-White F, Orsini A, Thomas S, Jayapal S, Trump N, Zaiwalla Z, Jayawant S. Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur J Paediatr Neurol. 2016;20(5):761–5.
Takahashi S, Yamamoto S, Okayama A, Araki A, Saitsu H, Matsumoto N, Azuma H. Electroclinical features of epileptic encephalopathy caused by SCN8A mutation. Pediatr Int. 2015;57(4):758–62.
Estacion M, O'Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, Meisler MH. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis. 2014;69:117–23.
de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM. van T SR, van Lieshout S, Nijman IJ, O'Brien JE, Hammer MF, Estacion M et al.: Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res. 2014;108(9):1511–8.
Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans MC, Kleefstra T, van de Warrenburg BP, Praamstra P, Nicolai J, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet. 2015;52(5):330–7.
Vaher U, Noukas M, Nikopensius T, Kals M, Annilo T, Nelis M, Ounap K, Reimand T, Talvik I, Ilves P, et al. De novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol. 2014;29(12):P202–6.
Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56(5):707–16.
Ohba C, Kato M, Takahashi S, Lerman-Sagie T, Lev D, Terashima H, Kubota M, Kawawaki H, Matsufuji M, Kojima Y, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55(7):994–1000.
Singh R, Jayapal S, Goyal S, Jungbluth H, Lascelles K. Early-onset movement disorder and epileptic encephalopathy due to de novo dominant SCN8A mutation. Seizure. 2015;26:69–71.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–30.
Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, Marcadier J, Sawyer SL, Mosca SJ, Innes AM, et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet. 2015;88(1):34–40.
Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217–21.
Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, Palmer R, Morrogh D, Boustred C, Hurst JA, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;
O'Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013;4:213.
Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK, Meisler MH. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2016;3(2):114–23.
Moller RS, Johannesen KM. Precision Medicine: SCN8A Encephalopathy Treated with Sodium Channel Blockers. Neurotherapeutics. 2016;13(1):190–1.
Boerma RS, Braun KP, van den Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, Lindhout D, van Kempen M, Boon M, Nicolai J, et al. Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics. 2016;13(1):192–7.
Acknowledgements
We would like to thank the patients and their family for their cooperation in this study.
Funding
The study was funded by 985 Peking University and Clinical Hospital Cooperation Project (2013-1-06), and Technology innovation talents special fund of Harbin Science and Technology Bureau (2016RAXYJ089).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
XHB, LPW, XRW, and SJY obtained the funding and design the research. QPZ, JRL and YC acquired and provided clinical data and samples from patient. HG performed the Whole-exome sequencing, Targeted NGS, PCR-Sanger sequencing and part of data analysis. JPW analyzed most of the data and draft the manuscript. XHB revised the manuscript. English language polishing was done by HG. All authors have read and approved the manuscript, and there are no conflicts of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by Clinical Research Ethics Committee, Peking University (NM: IRB00001052-11087). Written informed consent was obtained from all patients or their parents.
Consent for publication
Consent for publication (including medical data, and images) was obtained from the parents of all patients in this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The record of seizures attack of patients with SCN8A mutations. This file included the seizure frequency of patients with SCN8A mutations, at different periods. (DOCX 16 kb)
Additional file 2:
485 genes in the targeted-next generation sequencing panel. (PDF 45 kb)
Additional file 3:
23 candidate genes identified in the Chinese family with epilepsy, by whole-exome sequencing. (DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, J., Gao, H., Bao, X. et al. SCN8A mutations in Chinese patients with early onset epileptic encephalopathy and benign infantile seizures. BMC Med Genet 18, 104 (2017). https://doi.org/10.1186/s12881-017-0460-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0460-1