
Abstract
Background
Many osteoarthritis (OA) susceptibility genes have been identified in recent years. Given the overlap in the phenotype of joint inflammation between OA and Kashin-Beck disease (KBD), the aim of this study is to explore whether the reported OA susceptibility genes and two genes that may link to OA pathophysiology are associated with KBD in the Tibetan population.
Method
Fifteen single-nucleotide polymorphisms (SNPs) in 12 candidate genes previously reported as OA susceptibility loci were selected for investigation. Genotyping was performed using the SNaPshot method for these SNPs in a Tibetan population composed of 849 KBD patients and 565 normal controls. Meanwhile, the coding regions of two genes, COL10A1 and HABP2, which may involve in the pathological mechanism of OA/KBD, were sequenced by Sanger sequencing to identify susceptibility coding variants for KBD in the Tibetan population.
Results
The two arthritis-susceptible candidate SNPs, rs7775 (p.Arg324Gly) in the FRZB gene and rs7033979 in the ASPN gene, showed associations with KBD (OR = 1.568, P = 4 × 10−3 and OR = 0.744, P = 8 × 10−3, respectively). The coding variants rs142463796 (p.Asp128Asn) and rs2228547 (p.Gly545Arg) in the COL10A1 gene (OR = 9.832 and P = 6 × 10−3 and OR = 1.242, P = 0.043, respectively) and rs548354451 (p.Asp272Glu) in the HABP2 gene (OR = 2.813, P = 0.010) were associated with KBD patients.
Conclusion
These finding suggested that rs7775 in the FRZB gene may increase susceptibility to KBD, while rs7033979 in the ASPN gene may play a protective role in susceptibility to KBD in Tibetans. Moreover, genetic variants in chondrogenesis-related genes COL10A1 and HABP2 may play a role in the risk of developing KBD in the Tibetan population.
Similar content being viewed by others


Background
Kashin-Beck disease (KBD) was first hinted as an endemic blight in 1849 by a Russian surveyor who noted that people in villages along the Urov River suffered bone deformities. A few years later, a Cossack doctor, Nikolai Kashin, described Urov disease [1]. In 1906, another Cossack doctor, Evgeny Beck, documented cases in the monograph Osteoarthritis Deformans Endemica. After that, this type of joint deformity was called “Kashin-Beck Disease” [2]. More than 30 million people live in the endemic areas (northeastern to southwestern China, extending to southeastern Siberia and North Korea) [1]. China is the country with the largest incidence of KBD in the world, and the Tibetans are the people most affected by KBD in China. The prevalence of KBD in Tibetans peaked in the late 1950s, when in many severely hit villages, 60–90% of children showed signs of KBD [3, 4]. The incidence has declined steadily since 2000 due to a massive effort launched to stamp out KBD, including relocating populations to a non-endemic region, but in 2013, there remained 0.64 million patients with KBD and 1.16 million at risk in 377 counties of 13 provinces or autonomous regions in China [5].
KBD is a chronic osteoarthropathy combined with disturbances of flexion and extension in the ankles, knees, wrists and elbows, enlarged inter-phalangeal joints et al. Severely affected cases are characterized by disproportionate, stunted growth with associated joint deformity. The basic pathologic feature of KBD is the death of cartilage cells in the growth plate and articular surface, but its etiology has not been fully defined yet. The general presumption is that risk factors for this disease are nutritional deficiency of selenium and iodine and environmental contamination with mycotoxins and fulvic acid (FA). Selenium and/or iodine have been considered the most important deficiencies associated with KBD [6, 7]. Mycotoxins produced by fungi can contaminate grain, which may cause KBD, because mycotoxins cause lesions in cartilage tissues, especially in physeal cartilage [8]. FA as an exogenous free radical carrier, may accumulate on cartilage cells and lead to severe cartilage damage. Thus, high concentrations of FA in drinking water also have been implicated in the disease [9]. Cold and hypoxia also may be precipitating factors for KBD, consistent with the fact that all KBD-endemic regions are cold and/or hypoxic.
An increasing body of evidence suggests that environmental factors alone cannot account for the etiology of KBD [10]. In the hunt for genetic suspects, several studies have shown that genetic factors play an important role in KBD pathogenesis. In 2010, Xiong et al. showed that the polymorphism (rs1050450) of selenoprotein gene GPX1 was significantly associated with KBD in a Han Chinese population [11]. However, in our previous study of the Tibetan population, the single single-nucleotide polymorphism (SNP) (rs1050450) of the GPX1 gene was not significantly associated with KBD. Furthermore, haplotype analysis of SNPs rs1050450, rs1800668 and rs3811699 in the GPX1 gene showed significant association with KBD [7]. These results suggested that the GPX1 gene might play a protective role in susceptibility to KBD. In 2011, we reported that four SNPs (rs6457617, rs6457620, rs9275295 and rs7745040) in HLA-DRB1 gene locus were associated with KBD in the Tibetan population [12]. This result was confirmed in a different population by an independent group, revealing that two polymorphisms (rs7745040 and rs9275295) in the HLA-DRB1 gene and one polymorphism (rs9473132) in CD2AP gene have a significant statistical association with KBD [13]. Most recently, ITPR2 and COL9A1 were identified as susceptibility genes for KBD in Han Chinese [14, 15].
These studies suggested that genetic variants played an important role in KBD. Given that KBD has been found to have overlapping phenotypes and pathologic changes with osteoarthritis (OA), we suspected that OA-related gene variants might play an important role in the etiology and pathogenesis of KBD, so we analyzed the association of 15 SNPs in 12 OA-related genes with KBD in a Tibetan population.
In addition, the genes that code for structural proteins of the extracellular matrix (ECM) of the cartilage may play an important role in the development of KBD, especially the COL10A1 gene, which codes for collagen type X. COL10A1 has been proved to be a direct transcriptional target of RUNX2 during chondrogenesis. Immunohistological studies localized type X collagen exclusively in the zone of hypertrophic and calcifying cartilage [16]. And it was reported that Col10a1-Runx2 transgenic mice with delayed chondrocyte maturation were less susceptible to developing osteoarthritis [17]. Possible involvement of the HABP2 gene in KBD was suggested based on the following concepts. The HABP2 gene encodes an extracellular serine protease that binds to hyaluronic acid (also known as hyaluronan, HA). Both HA and HA binding protein (HABP) are important components of articular cartilage whose damage is the principal pathologic feature of KBD [18]. Intra-articular hyaluronic acid (IAHA) injection has been used to treat patients with KBD [19]. Therefore, we also investigated the potential association between KBD susceptibility and variants in COL10A1 and HABP2 genes by directly sequencing the exons of both genes.
Methods
Subjects and clinical examination
Approval for the study was provided by the Institutional Review Boards of Sichuan Provincial People’s Hospital. KBD patients and controls in this study were recruited from the Tibetan population in the same endemic villages in SongPan, RuoErGai and HongYuan counties in the Aba Tibetan Autonomous prefecture of Sichuan Province, China. All subjects signed informed consent forms prior to participation in the study.
KBD was diagnosed by physical examination and X-ray radiographs when a subject was more than 5 years old, had persistent pain, limitation of mobility and deformity of the shoulders, wrists, fingers, toes, ankles and knees [20], but did not have other types of arthritis, such as rheumatoid arthritis (RA) and OA, or local inflammation and a history of trauma [1, 6, 21]. The diagnosis of KBD was made in accordance with the KBD Chinese diagnosis criteria (GB16003-1995). Anteroposterior radiograph of the right hand of each patient was first taken with portable x-ray equipment. If the patient was classified as the first, second or third stage of KBD the anteroposterior and the lateral radiograph of the elbow, knee, ankle, hip were added. The subject had to have at least one of the following radiological signs was included: irregular erosions of the carpal/tarsal bones or the metacarpal/metatarsal bones and phalanges, irregular metaphyseal widening, cone-shaped epiphyses of phalanges [6]. Controls were individuals from the same regions with normal joint examinations and no other bone or joint diseases.
Selection of candidate genes and single-nucleotide polymorphism (SNP)
Selection of candidate genes was orientated toward the search for alterations in a gene that might be related to KBD and chondrodysplasia based on its function and possible pathological role. SNPs were selected for genotyping if they were considered to have confirmed associations or demonstrated suggestive evidence of association with OA in previous studies.
Genotyping
Venous blood was obtained from each subject and collected in an EDTA tube. Genomic DNA was purified using a Gentra Puregene Blood DNA kit (Minneapolis, MN). Genotyping was performed by the dye terminator-based SNaPshot method (Applied Biosystems, Foster City, CA). Additional file 1: Table S1 lists PCR and SNaPshot-specific primers. SNP analysis was performed on the ABI 3730 DNA analyzer (Applied Biosystems). Genotypes of the SNPs were determined by Gene mapper software (Applied Biosystems).
Exon sequencing
Primers were designed using Exon Primer (http://ihg.gsf.de/ihg/ExonPrimer.html) to amplify the exons of genes and at least 50 bp flanking the boundary of intron-exon junctions of each exon (Additional file 2: Table S2). Amplified exons then were sequenced using the ABI Big Dye on the ABI3730 DNA analyzer.
Statistical analysis
The Hardy-Weinberg equilibrium (HWE) of each SNP was tested by the goodness-of-fit χ2 test to compare the expected frequencies of genotypes in controls, and SNPs with P-values > 0.01 were considered to be within the HWE [22]. P values of the SNPs were calculated using an additive model. Odds ratios of the alleles and genotypes were estimated by the χ2 test. P-values < 0.05 were considered statistically significant. All statistical analyses were performed using SPSS version 17.0 software.
Results
Clinical features of the study subjects
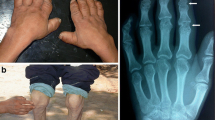
The study included 849 KBD patients and 565 controls. Table 1 lists information about patients and controls. There was no significant difference in gender distribution between KBD patients and controls (χ2 = 3.51, P > 0.05). KBD patients ranged from 5 to 75 years old, and the mean age was 53.7 ± 9.8 years for males and 53.5 ± 10.5 years for females. Controls ranged from 40 to 72 years old, with mean ages of 54.6 ± 10.0 years for males and 54.0 ± 9.5 years for females. As shown in Fig. 1a–c), the main features of KBD patients include flexion and extension in the knees, ankles, wrists and elbows and/or enlarged inter-phalangeal joints and so on, with the possibility of OA and/or RA being ruled out. No signs of KBD, OA or other autoimmune disease were detected in the controls.

Clinical features of patients with Kashin-Beck disease (KBD). a and b A patient with KBD shows obvious flexion and extension in the knees (circled area) and inter-phalangeal joints (arrow and angle labeled area), with deformity and limited mobility. c The x-ray radiographs of a KBD patient with irregular, enlarged hand inter-phalangeal joints
Association of SNPs in FRZB and ASPN genes with KBD
This study selected 15 SNPs (mapping to 12 genes) reported previously as suggestive or confirmed susceptibility loci for OA (Table 2) to investigate whether they were susceptibility loci for KBD in the Tibetan population. All 15 SNPs had a genotyping success rate > 96% and accuracy as judged by random re-genotyping of 10% of the samples by direct sequencing analysis. Table 3 presents the case–control comparison of allele frequencies for the 15 SNPs. Significant differences in the minor allele frequencies between the KBD cases and controls were detected at rs7775 of the FRZB gene and rs7033979 of the ASPN gene. Results show that the frequency of the minor allele “G” of rs7775 (p.Arg324Gly) in the FRZB gene was higher in patients with KBD than in controls (OR = 1.568, 95% CI = 1.151–2.136, P = 4 × 10−3). The frequency of the minor allele “G” of rs7033979 in the intron region of the ASPN (NM_001012267.2) gene was lower in patients with KBD than in controls (OR = 0.744, 95% CI = 0.597–0.927, P = 8 × 10−3). The other 13 OA-related SNPs were not significantly associated with KBD in the population used in this study, but we cannot exclude the possibility that these genes are involved in susceptibility to KBD. Because for most of these genes, only one polymorphism was studied in this study.
Association of variants in the COL10A1 and HABP2 genes with KBD
We carried out Sanger sequencing analyses of COL10A1 and HABP2 gene exons with the goal of identifying coding variants that may contribute to KBD risk in the Tibetan population using 849 KBD patients and 565 normal controls. In both KBD patients and controls, genotype distribution did not deviate significantly from the HWE (P_HWE > 0.05). In all, 21 and 23 non-synonymous changes were detected in COL10A1 and HABP2 genes, respectively (Additional file 3: Table S3). We found three of these variants, rs142463796 (p.Asp128Asn) and rs2228547 (p.Gly545Arg) in the COL10A1 gene and rs548354451 (p.Asp272Glu) in the HABP2 gene, were weakly associated with KBD (Table 4). The candidacy of rs142463796 in the COL10A1 gene is particularly strong given the low P value (OR = 9.832, 95% CI = 1.302–74.266, P = 6 × 10−3), in which the effect is associated with the minor allele “T” (causing aspartic acid to asparagine at amino acid 128 in the COL10A1 gene). SNP rs2228547 demonstrated a nominally significant association with KBD in the allelic tests (OR = 1.242, 95% CI = 0.999–1.544, P = 0.043). The frequency of the minor allele “A” of rs548354451 in the HABP2 gene differed between KBD patients and controls (OR = 2.813, 95% CI = 1.237–6.397, P = 0.010), and this variant led to the replacement of an aspartic acid by a glutamic acid at codon 272 (NP_004123).
Discussion
Although the cause of KBD remains elusive, it may well result from an interaction between genes and environment, which is supported by the following facts. First, moving populations from affected areas to unaffected areas significantly decreased disease prevalence, but KBD could not be abolished completely in the same population [12]. Second, although supplying selenium and/or iodine has produced positive effects in most KBD-affected areas, there are some affected areas where the effects of these supplements have been unclear [23]. Third, not everyone in the same family has KBD, even though they share the same environment, drink and eat the products in the KBD areas, including fungal toxins and are exposed to the same viruses et al [12]. Fourth, cattle in the affected areas do not show any KBD similar phenotypes.
Different genetic variants for each individual might explain why some populations are particularly vulnerable to KBD. KBD and OA have similar clinical manifestations and pathologic changes in the articular cartilage, such as chondrocyte necrosis and apoptosis, matrix degradation and cartilage degeneration [24]. Thus, KBD and OA may share some disease-related genes and pathogenesis. In this study, we investigated whether the 15 SNPs in 12 osteoarthritis-related genes are associated with KBD in a Tibetan population. We found that the SNPs rs7775 in the FRZB gene and rs7033979 in the ASPN gene showed significant associations with KBD.
The minor allele “G” of rs7775 in the FRZB gene was more common in patients with KBD than in controls (OR = 1.568, P = 4 × 10−3), suggesting that rs7775 might increase susceptibility to KBD. The FRZB gene encodes a soluble protein FRZB (also called sFRP-3), which is involved in negative regulation of the WNT signaling pathway. WNT/β-catenin signaling is a powerful stimulator of chondrocyte matrix catabolic action and may be part of mechanisms leading to excessive remodeling and degradation of cartilage matrix in joint pathologies [25]. Frzb (−/−) mice have been shown to contribute to cartilage damage by increasing WNT signaling and expression and activity of matrix metalloproteinase 3 (MMP-3) [26]. Several studies have explored the relationship between OA and rs7775 in the FRZB gene. By linkage and association studies, Loughlin et al. found that a p.Arg324Gly (rs7775) substitution in the FRZB gene was associated with hip OA in females (n = 338; P = 0.04) [27]. In a Dutch study, the rs7775 (p.Arg324Gly) was found to be associated with generalized osteoarthritis (GOA). Our data suggested for the first time that the rs7775 polymorphism was associated with KBD in the Tibetan population. Asporin (ASPN) is an extracellular matrix (ECM) protein that can bind to TGF-β1 and sequentially inhibit TGF-β/Smad signaling. TGF-β and BMP2 are crucial to differentiation and proliferation of perichondrial cells [28]. ASPN has been found expressed in high proportions in the cartilage of osteoarthritic patients. It acts as a negative regulator of chondrogenesis by inhibiting the action of TGF-β [29]. A meta-analysis conducted by Valdes et al. suggested that an ASPN allele, D13, is protective against the risk of knee OA in Japanese and Caucasians [30]. In this study, the frequency of the minor allele “G” of rs7033979 in the ASPN gene was less in KBD patients than in controls (0.157 versus 0.200), suggesting a protective effect of this variant on KBD (OR = 0.744, P = 4 × 10−3).
In the COL10A1 and HABP2 genes’ exon sequencing analysis, we found that two coding variants in the COL10A1 gene, rs142463796 and rs2228547, were significantly associated with KBD in the Tibetan population studied. The risk allele “T” of rs142463796 (p.Asp128Asn) had a 9.832-fold increased likelihood of developing KBD. COL10A1 codes for abundant proteins in the extracellular matrix. Abnormal COL10A1 expression and diverse mutations have been observed in multiple skeletal diseases, such as Cleidocranial Dysplasia (CCD) [31], SMCD [32] and SMD [33], implying a role of COL10A1 variants in KBD.
Based on the clinicopathologic role of cartilage cells in KBD patients, we supposed genes involved in extracellular matrix homeostasis in articular cartilage would play an important role in the development of KBD. We explored for the first time the association of variants in the HABP2 gene with KBD. We noted that the rare variant rs548354451 in the HABP2 gene was associated with KBD (OR = 2.813, P = 0.010). This variant led to the replacement of an aspartic acid by a glutamic acid at codon 272 (p.Asp272Glu), which is involved in a highly evolutionarily conserved residue in Euteleostomi from H.sapiens to M.musculus (Fig. 2). The protein encoded by the HABP2 gene is an extracellular serine protease that binds to hyaluronic acid, whose balance is important in maintaining the structure of cartilage and bone formation and, consequently, has been used to treat KBD patients [19].

Protein sequence alignment of human HABP2 with its orthologs. The residue of the missense mutation p.Asp272Glu (p.D272E) is highly conserved in Euteleostomi from H.sapiens to M.musculus
Overall, we genotyped 15 SNPs in12 candidate genes in Tibetan KBD patients and controls, as well as two genes derived from cartilage-related pathways. Our finding suggested that rs7775 in the FRZB gene might increase susceptibility to KBD, while rs7033979 plays a protective role in susceptibility to KBD in the endemic region. Furthermore, genetic variants in the chondrogenesis-related genes COL10A1 and HABP2 may play an important role in KBD pathogenesis. The fact that significant differences in genetic variants between cases and controls did not exist after Bonferroni correction (P = 0.05/tested loci number) could be explained by the relatively small sample size, with small effects of the variants. Further studies with larger cohorts are needed to confirm the findings of this study.
Conclusion
In this study, we reported two SNPs of osteoarthritis-related genes, rs7775 in the FRZB gene and rs7033979 in the ASPN gene, showed significant associations with KBD in Tibetans. These findings supporting the possibility of shared genetics etiology between OA and KBD. Moreover, we found that genetic variants in chondrogenesis-related genes COL10A1 and HABP2 may play a role in the risk of developing KBD in the Tibetan population.
Abbreviations
- ASPN:
-
Asporin
- CCD:
-
Cleidocranial Dysplasia
- COL10A1:
-
Collagen Type X Alpha 1 Chain
- FA:
-
Fulvic acid
- FRZB:
-
Frizzled-related protein
- GPX1:
-
Glutathione peroxidase 1
- HABP2:
-
Hyaluronan binding protein 2
- HLA-DRB1:
-
Major histocompatibility complex, class II, DR Beta 1
- HWE:
-
Hardy-Weinberg equilibrium
- KBD:
-
Kashin-Beck disease
- OA:
-
Osteoarthritis
- RA:
-
Rheumatoid arthritis
- SMCD:
-
Schmid-type metaphyseal chondrodysplasia
- SMD:
-
Japanese-type spondylometaphyseal dysplasia
- SNP:
-
Single-nucleotide polymorphisms
References
Hinsenkamp M. Kashin-beck disease. Int Orthop. 2001;25(3):133.
Stone R. A medical mystery in middle China. Science. 2009;324(5933):1378–81.
Mathieu F, Begaux F, Lan ZY, Suetens C, Hinsenkamp M. Clinical manifestations of Kashin-Beck disease in Nyemo valley, Tibet. Int Orthop. 1997;21(3):151–6.
Malaisse F, Mathieu F. Big bone disease: A multidisciplinary approach of Kashin-Beck disease in Tibet autonomous region (PR China). Gembloux (Belgique): Presses Agronomiques de Gembloux; 2008.
Guo X, Ma WJ, Zhang F, Ren FL, Qu CJ, Lammi MJ. Recent advances in the research of an endemic osteochondropathy in China: Kashin-Beck disease. Osteoarthr Cartil. 2014;22(11):1774–83.
Moreno-Reyes R, Suetens C, Mathieu F, Begaux F, Zhu D, Rivera MT, Boelaert M, Neve J, Perlmutter N, Vanderpas J. Kashin-Beck osteoarthropathy in rural Tibet in relation to selenium and iodine status. N Engl J Med. 1998;339(16):1112–20.
Huang L, Shi Y, Lu F, Zheng H, Liu X, Gong B, Yang J, Lin Y, Cheng J, Ma S, et al. Association study of polymorphisms in selenoprotein genes and Kashin-Beck disease and serum selenium/iodine concentration in a Tibetan population. PLoS One. 2013;8(8):e71411.
Chasseur C, Suetens C, Nolard N, Begaux F, Haubruge E. Fungal contamination in barley and Kashin-Beck disease in Tibet. Lancet. 1997;350(9084):1074.
Yang C, Niu C, Bodo M, Gabriel E, Notbohm H, Wolf E, Muller PK. Fulvic acid supplementation and selenium deficiency disturb the structural integrity of mouse skeletal tissue. An animal model to study the molecular defects of Kashin-Beck disease. Biochem J. 1993;289(Pt 3):829–35.
Lu AL, Guo X, Aisha MM, Shi XW, Zhang Y, Zhang YY. Kashin-Beck disease and Sayiwak disease in China: prevalence and a comparison of the clinical manifestations, familial aggregation, and heritability. Bone. 2011;48(2):347–53.
Xiong YM, Mo XY, Zou XZ, Song RX, Sun WY, Lu W, Chen Q, Yu YX, Zang WJ. Association study between polymorphisms in selenoprotein genes and susceptibility to Kashin-Beck disease. Osteoarthr Cartil. 2010;18(6):817–24.
Shi Y, Lu F, Liu X, Wang Y, Huang L, Long W, Lv B, Zhang K, Ma S, Lin H, et al. Genetic variants in the HLA-DRB1 gene are associated with Kashin-Beck disease in the Tibetan population. Arthritis Rheum. 2011;63(11):3408–16.
Yang Z, Xu Y, Luo H, Ma X, Wang Q, Wang Y, Deng W, Jiang T, Sun G, He T, et al. Whole-exome sequencing for the identification of susceptibility genes of Kashin-Beck disease. PLoS One. 2014;9(4):e92298.
Zhang F, Wen Y, Guo X, Zhang Y, Wang X, Yang T, Shen H, Chen X, Tian Q, Deng HW. Genome-wide association study identifies ITPR2 as a susceptibility gene for Kashin-Beck disease in Han Chinese. Arthritis Rheumatol (Hoboken, NJ). 2015;67(1):176–81.
Shi X, Zhang F, Lv A, Wen Y, Guo X. COL9A1 gene polymorphism is associated with Kashin-Beck disease in a northwest Chinese Han population. PLoS One. 2015;10(3):e0120365.
Kirsch T, von der Mark K. Isolation of human type X collagen and immunolocalization in fetal human cartilage. Eur J Biochem. 1991;196(3):575–80.
Lu Y, Ding M, Li N, Wang Q, Li J, Li X, Gu J, Im HJ, Lei G, Zheng Q. Col10a1-Runx2 transgenic mice with delayed chondrocyte maturation are less susceptible to developing osteoarthritis. Am J Transl Res. 2014;6(6):736–45.
Cao J, Li S, Shi Z, Yue Y, Sun J, Chen J, Fu Q, Hughes CE, Caterson B. Articular cartilage metabolism in patients with Kashin-Beck disease: an endemic osteoarthropathy in China. Osteoarthr Cartil. 2008;16(6):680–8.
Yu FF, Xia CT, Fang H, Han J, Younus MI, Guo X. Evaluation of the therapeutic effect of treatment with intra-articular hyaluronic acid in knees for Kashin-Beck disease: a meta-analysis. Osteoarthr Cartil. 2014;22(6):718–25.
Wang Y, Yang Z, Gilula LA, Zhu C. Kashin-Beck disease: radiographic appearance in the hands and wrists. Radiology. 1996;201(1):265–70.
Mathieu F, Begaux F, Suetens C, De Maertelaer V, Hinsenkamp M. Anthropometry and clinical features of Kashin-Beck disease in central Tibet. Int Orthop. 2001;25(3):138–41.
Shoemaker J, Painter I, Weir BS. A Bayesian characterization of Hardy-Weinberg disequilibrium. Genetics. 1998;149(4):2079–88.
Yao Y, Pei F, Kang P. Selenium, iodine, and the relation with Kashin-Beck disease. Nutrition (Burbank, Los Angeles County, Calif). 2011;27(11–12):1095–100.
Duan C, Guo X, Zhang X-D, Yu H-J, Yan H, Gao Y, Ma W-J, Gao Z-Q, Xu P, Lammi M. Comparative analysis of gene expression profiles between primary knee osteoarthritis and an osteoarthritis endemic to Northwestern China, Kashin-Beck disease. Arthritis Rheum. 2010;62(3):771–80.
Yuasa T, Otani T, Koike T, Iwamoto M, Enomoto-Iwamoto M. Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: its possible role in joint degeneration. Lab Investig. 2008;88(3):264–74.
Lories RJ, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, Thomas JT, Luyten FP. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007;56(12):4095–103.
Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, Ferreira A, Ciesielski C, Carson DA, Corr M. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A. 2004;101(26):9757–62.
Silverio-Ruiz KG, Martinez AE, Garlet GP, Barbosa CF, Silva JS, Cicarelli RM, Valentini SR, Abi-Rached RS, Junior CR. Opposite effects of bFGF and TGF-beta on collagen metabolism by human periodontal ligament fibroblasts. Cytokine. 2007;39(2):130–7.
Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, Mabuchi A, Kotani A, Kawakami A, Yamamoto S. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005;37(2):138–44.
Valdes AM, Loughlin J, Oene MV, Chapman K, Surdulescu GL, Doherty M, Spector TD. Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007;56(1):137–46.
Gu J, Lu Y, Li F, Qiao L, Wang Q, Li N, Borgia JA, Deng Y, Lei G, Zheng Q. Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell Death Dis. 2014;5:e1469.
Ikegawa S, Nakamura K, Nagano A, Haga N, Nakamura Y. Mutations in the N-terminal globular domain of the type X collagen gene (COL10A1) in patients with Schmid metaphyseal chondrodysplasia. Hum Mutat. 1997;9(2):131–5.
Ikegawa S, Nishimura G, Nagai T, Hasegawa T, Ohashi H, Nakamura Y. Mutation of the Type X Collagen Gene (COL10A1) Causes Spondylometaphyseal Dysplasia. Am J Hum Genet. 1998;63(6):1659–62.
Zhai G, van Meurs JB, Livshits G, Meulenbelt I, Valdes AM, Soranzo N, Hart D, Zhang F, Kato BS, Richards JB, et al. A genome-wide association study suggests that a locus within the ataxin 2 binding protein 1 gene is associated with hand osteoarthritis: the Treat-OA consortium. J Med Genet. 2009;46(9):614–6.
Hämäläinen S, Solovieva S, Vehmas T, Luoma K, Leino-Arjas P, Hirvonen A. Genetic influences on hand osteoarthritis in Finnish women–a replication study of candidate genes. PLoS One. 2014;9(5):e97417.
Shin MH, Lee SJ, Kee SJ, Song SK, Kweon SS, Park DJ, Park YW, Lee SS, Kim TJ. Genetic association analysis of GDF5 and ADAM12 for knee osteoarthritis. Joint Bone Spine. 2012;79(5):488–91.
Kerna I, Kisand K, Tamm A, Lintrop M, Veske K, Tamm A. Missense single nucleotide polymorphism of the ADAM12 gene is associated with radiographic knee osteoarthritis in middle-aged Estonian cohort. Osteoarthr Cartil. 2009;17(8):1093–8.
Kerna I, Kisand K, Tamm AE, Kumm J, Tamm AO. Two single-nucleotide polymorphisms in ADAM12 gene are associated with early and late radiographic knee osteoarthritis in Estonian population. Arthritis. 2013;2013:878126.
Bijsterbosch J, Kloppenburg M, Reijnierse M, Rosendaal FR, Huizinga TW, Slagboom PE, Meulenbelt I. Association study of candidate genes for the progression of hand osteoarthritis. Osteoarthr Cartil. 2013;21(4):565–9.
Valdes AM, Styrkarsdottir U, Doherty M, Morris DL, Mangino M, Tamm A, Doherty SA, Kisand K, Kerna I, Tamm A. Large scale replication study of the association between HLA class II/BTNL2 variants and osteoarthritis of the knee in European-descent populations. PLoS One. 2011;6(8):e23371.
Kerkhof HJ, Lories RJ, Meulenbelt I, Jonsdottir I, Valdes AM, Arp P, Ingvarsson T, Jhamai M, Jonsson H, Stolk L. A genome‐wide association study identifies an osteoarthritis susceptibility locus on chromosome 7q22. Arthritis Rheum. 2010;62(2):499–510.
Chapman K, Valdes AM. Genetic factors in OA pathogenesis. Bone. 2012;51(2):258–64.
Evangelou E, Valdes AM, Kerkhof HJ, Styrkarsdottir U, Zhu Y, Meulenbelt I, Lories RJ, Karassa FB, Tylzanowski P, Bos SD, et al. Meta-analysis of genome-wide association studies confirms a susceptibility locus for knee osteoarthritis on chromosome 7q22. Ann Rheum Dis. 2011;70(2):349–55.
Rodriguez-Lopez J, Pombo-Suarez M, Liz M, Gomez-Reino JJ, Gonzalez A. Further evidence of the role of frizzled-related protein gene polymorphisms in osteoarthritis. Ann Rheum Dis. 2007;66(8):1052–5.
Meulenbelt I, Seymour AB, Nieuwland M, Huizinga TW, van Duijn CM, Slagboom PE. Association of the interleukin‐1 gene cluster with radiographic signs of osteoarthritis of the hip. Arthritis Rheum. 2004;50(4):1179–86.
Loughlin J, Mustafa Z, Dowling B, Southam L, Marcelline L, Raeinae SS, Ala-Kokko L, Chapman K. Finer linkage mapping of a primary hip osteoarthritis susceptibility locus on chromosome 6. Eur J Hum Genet. 2002;10(9):562–8.
Loughlin J, Meulenbelt I, Min J, Mustafa Z, Sinsheimer JS, Carr A, Slagboom PE. Genetic association analysis of RHOB and TXNDC3 in osteoarthritis. Am J Hum Genet. 2007;80(2):383.
Shi D, Nakamura T, Nakajima M, Dai J, Qin J, Ni H, Xu Y, Yao C, Wei J, Liu B. Association of single-nucleotide polymorphisms in RHOB and TXNDC3 with knee osteoarthritis susceptibility: two case–control studies in East Asian populations and a meta-analysis. Arthritis Res Ther. 2008;10(3):R54.
Valdes AM, Spector TD, Tamm A, Kisand K, Doherty SA, Dennison EM, Mangino M, Tamm A, Kerna I, Hart DJ. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010;62(8):2347–52.
Liying J, Yuchun T, Youcheng W, Yingchen W, Chunyu J, Yanling Y, Hongmei J, Yujie L. A SMAD3 gene polymorphism is related with osteoarthritis in a Northeast Chinese population. Rheumatol Int. 2013;33(7):1763–8.
Jones CA, London NR, Chen H, Park KW, Sauvaget D, Stockton RA, Wythe JD, Suh W, Larrieu-Lahargue F, Mukouyama YS, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med. 2008;14(4):448–53.
Acknowledgements
We would like to thank all the KBD patients, controls and their families for participating in this study.
Funding
This research project was supported by: the National Natural Science Foundation of China (81170883 (Z.Y.), 81300802 (L.H.) and 81170882 (Y.S)); and the Department of Science and Technology of Sichuan Province, China (2014SZ0169, 2015SZ0052 (Z.Y.), 2015JQO057 (L.H.), 2013JY0195 (L.H), 2015SZ0060 (Y.L.) and 2014JZ0004 (Y.S))
Availability of data and materials
All raw data generated or data analysed during this study are available from the corresponding author upon request.
Authors’ contributions
ZLY was responsible for concept and design, contributed to interpretation of results, the integrity of the data and the accuracy of the data analysis, and drafted and revised final manuscript. ZFT and LLH performed data analysis and interpretation of results, and drafted and revised final manuscript. YS, FL, XL, SM, JC, HL, YYL and ZLY was involved in the collaborative effort to collect biological samples. All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Consent was obtained from all participants for publication of this result and the accompanying images.
Ethics approval and consent to participate
Informed consent was obtained from all subjects involved in this study. Subjects were clinically diagnosed at the Sichuan Provincial People’s Hospital, China. All procedures were reviewed and approved by the ethics committee of the hospital.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primers of selected SNPs used in this study. (DOC 67 kb)
Additional file 2: Table S2.
Primers used for exons sequencing. (DOC 48 kb)
Additional file 3: Table S3.
Rare variants detected in the COL10A1 and HABP2 exon sequencing of KBD Cases and normal controls. (DOC 80 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tai, Z., Huang, L., Lu, F. et al. Association study of candidate genes for susceptibility to Kashin-Beck disease in a Tibetan population. BMC Med Genet 18, 69 (2017). https://doi.org/10.1186/s12881-017-0423-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0423-6