
Abstract
Background
Niemann-Pick disease type C (NPC) is an inherited metabolic disorder; due to defect in cellular cholesterol trafficking. It is clinically a heterogeneous disease with variable age of onset with multiple organ systems being involved. NPC1 gene is involved in 95% cases where as remaining ~5% cases are linked with NPC2 gene.
Case presentation
Case-1, a 14-months-old female presented with recurrent respiratory distress, failure to thrive and hepatosplenomegaly. Lung biopsy was suggestive of alveolar proteinosis and liver biopsy confirmed foamy macrophages. Molecular analysis revealed homozygous mutation c.141C > A in exon 2 of NPC2 gene.
Case-2, a 3-year-old male presented with dyspnoea and hepatomegaly noticed at 1 year of age. HRCT-scan of thoracic region showed consolidation with mediastinal lymphadenopathy. Broncho-alveolar lavage revealed moderate amount of foamy macrophages and bone marrow examination detected foam cells. Homozygous T > C transition in intron 1 of the NPC2 gene was identified.
Conclusion
Our study demonstrates that NPC2 can present in early years of life with pulmonary complications like alveolar proteinosis and hepatosplenomegaly or hepatomegaly due to mutation in NPC2 gene. An early suspicion will help clinicians to clinch its diagnosis, management and genetic counselling.
Similar content being viewed by others


Background
Niemann–Pick disease type C (NPC) is a fatal autosomal recessive neurovisceral disorder due to mutation in NPC1 and NPC2 genes leading to alterations in trafficking of endocytosed cholesterol [1]. Due to heterogeneous clinical phenotype, NPC is underdiagnosed and often missed altogether. In NPC, the protein product of the NPC1 gene functions as a transporter of cholesterol and glycolipids in the endosomal-lysosomal system whereas the smaller protein product of NPC2 cooperates with the NPC1 protein [2, 3]. NPC2 plays a vital role in endosomal/lysosomal cholesterol trafficking by markedly accelerating the rates of transport from and between membranes [4]. The mechanism of NPC2 action involves direct interaction of the protein with membranes. The defects in NPC1 and NPC2 proteins, leads to sequestration of cholesterol derived products in the cell leading to hepatosplenomegaly, pulmonary and neurological manifestations [3]. NPC1 gene is responsible for 95% of manifestations with main phenotype being hepatosplenomegaly and nervous system. The other ~5% of the NPC is caused by NPC2 gene where pulmonary manifestations with respiratory failure have been documented [5]. Till date nearly twenty cases have been reported of NPC2 with twelve homoallelic mutations and none from India. Present study is the first report of NPC2 from India with primary pulmonary manifestations and hepatosplenomegaly highlighting phenotype-genotype correlation.
Case presentation
Case-1: 9 month old female child born to non-consanguineous parents from Rajasthan (Marwadi community) presented with recurrent respiratory tract infection and failure to thrive. She was frequently hospitalized for tachypnea and developed hepatosplenomegaly in next 5 months. Multiple treatment modalities were tried including antibiotics, nebulisations, anti-fungal agents, systemic corticosteroids and moist oxygen inhalation. In spite of all these, child remained tachypneic and continued to be oxygen dependent.
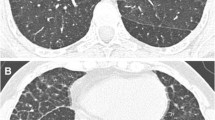
Her chest radiogram showed hazy, diffuse bilateral alveolar infiltrates. HRCT showed ground glass opacification of the right upper lobe, suggestive of alveolar proteinosis. Lung biopsy with PAS staining showed dilated alveoli, lined by type-2 pneumocytes, containing foamy macrophages further indicating alveolar proteinosis. Possibility of a secondary alveolar proteinosis was kept in view of hepatosplenomegaly along with lung involvement. Liver biopsy showed fairly well preserved lobular architecture and cord pattern, few lymphocytes in portal tract and finely vacuolated ballooned hepatocytes and kupffer cell were suggestive of Niemann-Pick disease. Further studies revealed high plasma chitotriosidase level with fifty percent reduced activity of acid Sphingomyelinase [3.6 nmol/hr/mg – NR: 8.0-14.5 nmol/hr/mg protein]. This has clinched the clinical suspicion of NPC in the proband. Infilatration of the lungs with liver disease in the proband pointed towards NPC2 diagnosis. Therefore bi-directional sanger sequencing covering exon-intron boundries of NPC2 gene was carried out. This has identified homozygous nonsense variant (c.141C > A) (p.Cys47Ter) in exon 2 (Fig. 1a). This variant is reported as pathogenic in HGMD (CM052291) and ClinVar (dbSNP80358263) database and responsible for protein truncation (variation ontology:0015).
Case-2: 1 year old male child born to third degree consanguineous parents from Maharshtra (Brahmin community) presented with dyspnoea, hepatomegaly and failure to thrive. Limbs were hypotonic with normal reflexes and power. Chest X-rays showed hyperinflation initially and progressive perihilar alveolar infiltration sparing peripheries. HRCT scan of thoracic region showed patchy alveolar fibrosis with mediastinal lymphadenopathy. Broncho-alveolar lavage revealed moderate amount of foamy macrophages and bone marrow examination showed presence of foamy histiocytic cells. Enzyme study for Niemann Pick type A, B and Gaucher’s disease were normal. Since acid sphingomyelinase was normal and pulmonary manifestations and hepatomegaly were predominantly present, NPC was suspected. Further study was carried out by bi-directional Sanger sequencing of NPC2 gene covering exon-intron boundries. This has identified homozygous splice site variation c.82 + 2 T > C (g.5191 T > C) in NPC2 gene (Fig. 1b). This variant is reported as pathogenic in HGMD (CS032706) and ClinVar (dbSNP:879253740) database that affect RNA splicing (variation ontology:0362).

Sequencing of the NPC genes. a Chromatogram showing homozygous nonsense mutation c.141C > A (p.C47X) in exon 2 of NPC2. b Homozygous intronic c.82 + 2 T > C transition mutation detection in intron 1 of NPC2
Discussion
NPC is a rare neurovisceral disease with an overall incidence of 1:150,000, of which only 5% cases are linked to NPC2 [6]. It is caused due to deposition of unesterified cholesterol mainly in brain, liver and lungs. Primary diagnosis of NPC by filipin stain is considered as the gold standard [7]. However, being tedious and time consuming, molecular testing was introduced in 2000 where the molecular defect was detected in NPC1 and NPC2 gene located on chromosome 18q11.2 and 14q24.3 respectively [3]. Since then several studies have shown an association of pulmonary manifestation and hepatosplenomegaly with NPC2 [8–11]. Likewise the two cases under study had a similar clinical presentation and were investigated primarily for acid sphingomyelinase (aSMase) activity in leucocytes. Case 1 had shown nearly 50% reduced activity of sphingomyelinase while case 2 had shown normal activity. Nearly fifty percent reduced activity of spingomyelinase in case 1 can be due to gross disruption of sphingomyelinase processing by intracellular cholesterol loading affecting aSMase activity as has been shown in CHO cell lines [12]. Pulmonary phenotype with reduced aSMase activity gave a clue of NPC2 in case 1. Identical phenotype led to consideration of NPC2 in the second case. Molecular study has identified previously known pathogenic non sense and RNA splicing mutations respectively. Both NPC2 mutations are known to be associated with pulmonary manifestations which may be a result of loss of normal NPC2 protein expression in alveolar macrophages leading to an accumulation of functionally inactive surfactant rich in cholesterol [11].
In absence of specific biomarkers for NPC2, non-enzymatic plasma levels of certain cholesterol oxidation products (oxysterols) can be used for clinical screening of NPC. However, this screening method alone cannot distinguish between causative gene NPC1 from NPC2 [13].
NPC1 and NPC2 carrying mutants are identical in terms of pathogenesis and clinical phenotype/s but there lies a discrete demarcation where pulmonary manifestations give clue to the diagnosis of NPC2 gene [14]. Identification of NPC2 as the causative factor for NPC with pulmonary involvement was shown by Park et al. for the first time in 2003 [15]. Moreover, three mutations in the NPC2 gene were subsequently notified by Verot et al. in 2007 [16]. In 2005, Chikh et al. demonstrated that missense mutation of NPC2 causes misfolding of endoplasmic reticulum proteins [17].
A detailed clinical analysis and mutation in NPC2 have clearly shown pronounced lung involvement as a cause of early death in six patients as shown by Millat et al. and several others [18–20]. Similar severity was observed in the present cases. Till date, nearly twenty patients with twelve different homoallelic mutations in NPC2 gene with pulmonary manifestations have been reported (Fig. 2, Table 1) [20, 21]. Documentation of these mutations with phenotypic presentation has become obligatory as commonly occurring bronchitis/pneumonia with liver involvement and failure to thrive are likely to mimic NPC2. Our study emphasizes the association of homoallelic mutation of NPC2 gene with non responsive alveolar fibrosis/proteinosis and hepatomegaly.

Schematic view of mutations reported in NPC2 gene having pulmonary manifestation. Mutations observed in the present study are shown in bold
Reporting more cases of NPC2 having pulmonary phenotype will help the clinicians to reach at an early diagnosis and counselling to the family for possible therapeutic options. Though Miglustat therapy is known to be useful for NPC disease with neurological disease but not with pulmonary manifestations [22] successful allogeneic bone marrow transplant [23] was performed in one patient with NPC2 and may be helpful in further patients once early diagnosis is clinched.
Conclusion
Mutations in NPC2 gene were studied for the first time in Indian subcontinent and rarely been reported in the literature. Accurate recognition of young patients with primary unexplained alveolar fibrosis/proteinosis together with hepatomegaly suggesting NPC with molecular characterization of NPC2 gene is essential. This will provide important new information about functional domains of NPC2 and advance the understanding of its precise function and its potential interactions with NPC1. Moreover, it will enhance our knowledge regarding the underlying pathogenic mechanism responsible for pulmonary manifestation.
Abbreviations
- HRCT-scan:
-
High Resolution Computer Tomography Scan
- NPC:
-
Niemann–Pick disease type C
- OPD:
-
Out Patient Department
- PAS:
-
Periodic Acid Schiff
References
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Cheruku SR, Xu Z, Dutia R, Lobel P, Storch J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal transport. J Biol Chem. 2006;281:31594–604.
Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–301.
Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A. 1985;82:8247–51.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. NP-C Guidelines Working Group.; Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–44.
Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann–Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917–20.
Sheth JJ, Sheth FJ, Oza N. Niemann-Pick type C disease. Indian Pediatr. 2008;45:505–7.
Vanier MT, Pentchev P, Rodriguez-Lafrasse C, Rousson R. Niemann-Pick disease type C: an update. J Inherit Metab Dis. 1991;14:580–95.
Liscum L, Sturley SL. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim Biophys Acta. 2004;1685:22–7.
Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–81.
Griese M, Brasch F, Aldana VR, Cabrera MM, Goelnitz U, Ikonen E, et al. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin Genet. 2010;77:119–30.
Reagen Jr JW, Hubbert ML, Shelness GS. Posttranslational regulation of acid sphingomyelinase in NPC-1 fibroblasts and free cholesterol enriched chinese hamster cells. J Biol Chem. 2000;275:38104–10.
Reunert J, Lotz-Havla AS, Polo G, Kannenberg F, Fobker M, Griese M, et al. Niemann-Pick Type C-2 Disease: Identification by Analysis of Plasma Cholestane-3β,5α,6β-Triol and Further Insight into the Clinical Phenotype. JIMD Rep. 2015;23:17–26.
Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, et al. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A. 2004;101:5886–91.
Park WD, O’Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS, et al. Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat. 2003;22:313–25.
Verot L, Chikh K, Freydiere E, Honore R, Vanier MT, Milla G. Niemann-Pick C disease: functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin Genet. 2007;71:320–30.
Chikh K, Rodriguez C, Vey S, Vanier MT, Millat G. Niemann-Pick type C disease: subcellular location and functional characterization of NPC2 proteins with naturally occurring missense mutations. Hum Mutat. 2005;26:20–8.
Millat G, Chikh K, Naureckiene S, Sleat DE, Fensom AH, Higaki K, et al. Niemann-Pick disease type C: spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am J Hum Genet. 2001;69:1013–21.
Schofer O, Mischo B, Püschel W, Harzer K, Vanier MT. Early-lethal pulmonary form of Niemann-Pick type C disease belonging to a second rare genetic complementation group. Eur J Pediatr. 1998;157:45–9.
Yaman A, Eminoğlu FT, Kendirli T, Ödek Ç, Ceylaner S, Kansu A, et al. A rare cause of fatal pulmonary alveolar proteinosis: Niemann-Pick disease type C2 and a novel mutation. J Pediatr Endocrinol Metab. 2015;28:1163–7.
Reunert J, Fobker M, Kannenberg F, Chesne ID, Plate M, Wellhausen J, Rust S, Marquardt T. Rapid diagnosis of 83 patients with Niemann Pick type C disease and related cholesterol transport disorders by cholestentriol screening. EBiomedicine. 2015. doi:10.1016/j.ebiom.2015.12.018.
Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 2009;98:243–9.
Bonney DK, O’Meara A, Shabani A, Imrie J, Bigger BW, Jones S, et al. Successful allogeneic bone marrow transplant for Niemann-Pick disease type C2 is likely to be associated with a severe ‘graft versus substrate’ effect. J Inherit Metab Dis. 2010;33 Suppl 3:S171–3.
Acknowledgments
We thank the patient and the parents for their cooperation.
Funding
This work is partly supported by intramural grant from FRIGE’s Institute of Human Genetics and Indian Council of Medical Research [grant no.: 54/01/2009-BMS]. None of the funding agencies were involved in the study design, specimen collection, analysis, interpretation of the data and preparation of the manuscript.
Availability of data and materials
The dataset supporting the conclusion of this article is available in the ClinVar repository
Clinvar submission accession ID for NPC2
SCV000272928.1 (p.Cys47Ter)
https://www.ncbi.nlm.nih.gov/clinvar/variation/21456/
SCV000272929.1 (c.82+2T>C)
https://www.ncbi.nlm.nih.gov/clinvar/variation/242903/
Authors’ contributions
Conceived and designed the experiments: JS and MM. Clinical analysis: JJ and MM. Enzyme and molecular analysis: JS and MM. Wrote the first draft of the manuscript: KS and FS. Made critical revisions and approved final version: KS, JS and FS. All authors reviewed and approved of the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Informed written consent was obtained from all the participants for publication of their clinical details and/or clinical images. A copy of the written consent is available for review by the editor of this journal.
Ethics approval and consent to participate
Present case under submission has been approved by the institutional ethics committee [FRIGE’s Institute of Human Genetics] wide approval number FRIGE/IEC/5/2010 dated 7th March, 2010. This process is in accordance with the Helsinki declaration.
An informed consent was obtained from the parents before enrolling for the investigations [This was in accordance with the requirement of the institutional ethics committee].
An informed consent for publication was also obtained from the individuals included in the submission [This was in accordance with the requirement of the institutional ethics committee].
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sheth, J., Joseph, J.J., Shah, K. et al. Pulmonary manifestations in Niemann-Pick type C disease with mutations in NPC2 gene: case report and review of literature. BMC Med Genet 18, 5 (2017). https://doi.org/10.1186/s12881-017-0367-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0367-x