
Abstract
Background
Congenital cataracts affect 3–6 per 10,000 live births and represent one of the leading causes of blindness in children. Congenital cataracts have a strong genetic component with high heterogeneity and variability.
Case presentation
Analysis of whole exome sequencing data in a patient affected with congenital cataracts identified a pathogenic deletion which was further defined by other techniques. A ~98-kb homozygous deletion of 6p24.3 involving the first three exons (two non-coding and one coding) of GCNT2 isoform A, the first exon (coding) of GCNT2 isoform B, and part of the intergenic region between GCNT2 and TFAP2A was identified in the patient and her brother while both parents were found to be heterozygous carriers of the deletion. The exact breakpoints were identified and revealed the presence of Alu elements at both sides of the deletion, thus indicating Alu-mediated non-homologous end-joining as the most plausible mechanism for this rearrangement. Recessive mutations in GCNT2 are known to cause an adult i blood group phenotype with congenital cataracts in some cases. The GCNT2 gene has three differentially expressed transcripts, with GCNT2B being the only isoform associated with lens function and GCNT2C being the only isoform expressed in red blood cells based on earlier studies; previously reported mutations/deletions have either affected all three isoforms (causing blood group and cataract phenotype) or the C isoform only (causing blood group phenotype only). Dominant mutations in TFAP2A are associated with syndromic anophthalmia/microphthalmia and other ocular phenotypes as part of Branchio-Ocular-Facial-Syndrome (BOFS). While the patients do not fit a diagnosis of BOFS, one sibling demonstrates mild overlap with the phenotypic spectrum, and therefore an effect of this deletion on the function of TFAP2A cannot be ruled out.
Conclusions
To the best of our knowledge, this is the first case reported in which disruption of the GCNT2 gene does not involve the C isoform. The congenital cataracts phenotype in the affected patients is consistent with the previously defined isoform-specific roles of this gene. The GCNT2-TFAP2A region may be prone to rearrangements through Alu-mediated non-homologous end-joining.
Similar content being viewed by others


Background
Congenital cataracts are diagnosed within the first year of life. These cataracts are one of the leading causes of blindness in children and are estimated to occur with a prevalence of 3–6 per 10,000 live births [1]. Congenital cataracts may appear either in isolation or in association with other ocular or systemic anomalies. Up to 25 % of congenital cataracts are thought to be caused by genetic defects [2]. The genetic landscape of mutations causing congenital cataract is extremely diverse; more than 40 genes and additional loci have been associated with nonsyndromic cataract [2–6].
GCNT2 (glucosaminyl (N-acetyl) transferase 2, I-branching enzyme) was first identified in 2001 as the gene encoding for the glycosyltransferase responsible for the human blood group I antigen. Recessive mutations in GCNT2 result in an adult i blood group phenotype, which is also associated with congenital cataracts in some cases [7]. Alternative splicing of the GCNT2 gene produces three transcripts (A, B, and C). The three transcripts share a common second and third coding exon with a unique first exon for each isoform; differing expression profiles were identified for the transcripts with only the GCNT2B isoform expressed in lens epithelial cells and only the GCNT2C isoform expressed in reticulocytes [8]. To date, seven missense mutations, one nonsense mutation, and two large deletions have been reported; mutations in exon 1C, affecting only the GCNT2C isoform, cause the adult i blood group without cataracts while mutations/deletions affecting exons 2 and 3, shared by all isoforms, result in the adult i blood group along with congenital cataract [7–11].
Case presentation
Patient 1(individual II:1) is an 18-month old Pakistani female affected with bilateral dense central congenital cataract (Fig. 1a, Table 1) which were visually significant and required extraction at 2 months of age, mild asymmetry of the palpebral fissures, and left nasolacrimal duct obstruction; her development is normal and growth parameters are generally normal with the exception of borderline microcephaly (length 83.8 cm, 75–90th centile; weight 10.2 kg, 25–50th centile; and head circumference 44 cm (3rd centile)). Physical exam at 4 months of age identified hypotelorism (familial) and mildly widely-spaced nipples. Her younger brother, age 6 months, was similarly affected with visually significant bilateral dense central congenital cataracts requiring extraction around 2 months of age; his length (67.5 cm, 25–50th centile), weight (6.8 cm, 5–10th centile), and head circumference (42.5 cm, 10–25th centile) are all within the normal range (Table 1). Family history shows unaffected second-cousin parents with additional endogamous mating within the family. A double second-cousin to the proband is affected with bilateral non-syndromic anophthalmia/ microphthalmia with no additional details available.

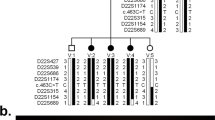
Patient photographs and pedigree. a Photograph of Patient 1’s eyes at 2 months of age showing bilateral cataract. b Pedigree showing both affected siblings with a homozygous deletion of 6p24.3 while the unaffected parents are heterozygous carriers. WT: wild type; black arrow indicates proband
Materials and methods
Whole exome sequencing was performed by Macrogen (previously Axeq) and analyzed as previously described [12]; briefly, exome data from the proband was analyzed using the SNP & Variation Suite (SVS; Golden Helix, Bozeman, MT, USA) to identify/exclude mutations in the coding and splicing regions of 40 known nonsyndromic cataract genes and 7 additional crystallins [3–6]; synonymous variants and variants with a frequency of >1 % in the general population (http://exac.broadinstitute.org, http://evs.gs.washington.edu/EVS/, http://www.1000genomes.org/) were considered to be benign variants. Copy number variation analysis was completed by screening exome sequencing data using the Copy Number Inference From Exome Reads (CoNIFER) v0.2.2 software package as previously outlined [13]; regions of interest were further verified by independent quantitative PCR reactions using DNA samples from the proband and other available familial samples with SYBR Green PCR Master Mix (Applied Biosystems/Life Technologies, Carlsbad, CA, USA). qPCR reactions utilized three region-specific probes (Additional file 1: Table S1) and were performed as follows: primers located within regions of interest were designed using Primer3Plus software (http://sourceforge.net/projects/primer3/) using qPCR settings. Each reaction was comprised of five nanograms of DNA in a total reaction volume of 12uL. Each primer set was run three times in triplicate using patient, parental or control DNA on a Bio-Rad CFX Connect Real-Time PCR machine (Bio-Rad, Hercules, CA, USA). A primer set for the housekeeping gene RPPH1 (ribonuclease P RNA component H1) was used to normalize all data. A probe located in NDP (Norrie disease (pseudoglioma)), located on the X-chromosome, was used as a copy-loss control. All experiments included a no-template control and an unaffected human DNA sample with presumably normal copy number at each region for comparison. Copy number changes were calculated using the 2-ΔΔCt method as previously described [14]. Following qPCR confirmation, the size and exact breakpoints of the deletion were determined using a series of regular PCR reactions that utilized primers located on both ends of the region (as defined by CoNIFER and qPCR analysis) and standard conditions (Additional file 1: Table S1). Since the patients were apparently homozygous for the deletion, no amplification product indicated that the primer(s) are located inside of the deleted region while the presence of a PCR product indicated primers outside of the deletion. Once sequences bordering the deleted region on the centromeric and telomeric sides were determined, the corresponding primers were used to amplify a 1.5 kb region across the breakpoints. The resultant product was cloned into pCRII-TOPO® (Life Technologies, Carlsbad, CA, USA) vector using the manufacturer’s protocols and sequenced bidirectionally with M13 forward and reverse primers using Big Dye Terminator v3 chemistry and an ABI 3730XL sequencer (Applied Biosystems/Life Technologies, Carlsbad, CA, USA); the obtained sequences were compared with the corresponding reference sequence using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results and discussion
Review of the whole exome sequencing (WES) data from Patient 1 did not identify any potentially pathogenic variants (with only two synonymous variants) in known nonsyndromic cataract genes. The WES data was then analyzed for copy number variation which revealed a potential 208-kb deletion (6p24.3 chr6: 10,412,788-10,621,660) affecting TFAP2A and GCNT2. The deletion was verified using qPCR probes located in the first coding exon of GCNT2 isoform A and the first exon of TFAP2A; the qPCR confirmed deletion of the GCNT2 sequence in both unaffected parents (haploid, heterozygous) and affected children (complete loss, homozygous) while diploid copy of the TFAP2A sequence was identified in all family members. Further analysis of the region by a series of regular PCR reactions using affected DNA identified the centromeric breakpoint between chr6:10472330–10472606 (set 7; diploid) and chr6:10474759–10474901 (set 8; complete loss) and the telomeric breakpoint between chr6:10570580–10570905 (set 13, complete loss) and chr6:10571951–10572257 (set 14, diploid). Primer sets designed to span the deleted region produced a ~1.5 kb product from the DNA of the affected patients. Sequencing of this product identified the exact deletion breakpoint sites: their analysis revealed the presence of Alu repeats and specifically a 12-bp identical sequence at both sides of the deleted region of 97.974-kb (hg19, chr6: 10,473,864–10,571,838) (Fig. 2). The homozygous deletion encompassed four exons of GCNT2 (the first two noncoding and one coding exons of isoform A and the first coding exon of isoform B) and extended 47.471-kb upstream of the most 5’ exon of the GCNT2 gene (Fig. 2). The distance from the telomeric end of the deletion to the nearest protein-coding gene, TFAP2A (transcription factor AP-2 alpha), is 54.300-kb. The distance from the centromeric end of the deletion to the fist exon of GCNT2 isoform C is 13.922-kb. Although GCNT2C and TFAP2A were not included in the deletion, effects on their expression through possible interference with regulatory elements cannot be ruled out.

Schematic presentation of the chromosome 6p24.3−24.2 region and the identified deletion. The UCSC Genome Browser (http://genome.ucsc.edu) view of the deleted region indicating the positions of genes is included; the deletion identified in the affected family is shown as a rectangular red box; the DNA sequence across the breakpoint for the deleted allele is shown at the bottom of the drawing with regions corresponding to the telomeric and centromeric flanks of the deletion indicated by dashed lines and a 12-nt repeat highlighted in red font
Genomic deletions of GCNT2 have been previously reported in two families with blood group i and congenital cataracts but both deletions included exons 2 and 3 which are shared by all isoforms [7, 11]. Borck and colleagues noted that the GCNT2 locus is rich with Alu elements and therefore is likely a hotspot for deletions or duplications to occur [11]. The GCNT2 gene has three differentially expressed transcripts, with GCNT2B being the only isoform associated with lens function and GCNT2C being the only isoform expressed in red blood cells [8]. The GCNT2 protein modifies the i antigen, a linear sphingoglycolipid present on the cell surface of most human cells as well as on glycoproteins in body fluids, into the active branched I antigen; the i/I antigens are thought to play a role in the regulation of cell growth and differentiation in the developing lens [8, 9].
The deletion described in this case report differs from previously reported deletions and mutations since it only affects the GCNT2A and GCNT2B isoforms and leaves the GCNT2C isoform intact. Previous studies demonstrated that only the GCNT2B isoform is expressed in lens epithelial cells and patients with mutations which specifically affect the C isoform demonstrate the adult i phenotype without congenital cataracts [8]. Thus, the presence of cataract in the affected patients reported here with a clear disruption of GCNT2A and B isoforms only is consistent with the isoform-specific roles identified for this gene. Additionally, in the case reported here we were able to identify the exact sequences at the breakpoints and clearly implicate Alu-mediated non-homologous end-joining as a mechanism for this rearrangement. This mechanism has been previously reported by our and other groups [15, 16]).
The deletion reported here also extends into the genomic region upstream of GCNT2 and TFAP2A which are positioned in a head-to-head orientation. TFAP2A is approximately 100-kb distal to GCNT2 and the deletion removes approximately 47-kb of genomic sequence between the two genes. It is possible that this deletion could affect TFAP2A function through removal/rearrangement of regulatory elements, as has been shown for other genes [17–19]. TFAP2A is a retinoic acid responsive transcription factor which is required for normal development of the lens and optic cup as well as for parts of the craniofacial region. Heterozygous mutations in TFAP2A cause Branchio-Ocular-Facial-Syndrome (BOFS) characterized by craniofacial phenotypes (distinct facial features, microcephaly, and cleft lip/palate), skin defects in the cervical region or regions around the ear, ocular defects (microphthalmia, coloboma, strabismus, cataract, or ptosis), lacrimal duct obstruction, and hearing loss [20–22]. Missense mutations account for the majority of TFAP2A variants, however whole gene deletions have also been reported. To date, no deletions affecting the upstream region of TFAP2A, but not the coding region itself, have been reported. Careful physical examination of the patients did not identify sufficient features to warrant a diagnosis of BOFS in the siblings, but Patient 1 did show borderline microcephaly, mild asymmetry of the palpebral fissures, left nasolacrimal duct obstruction, and somewhat widely spaced nipples. While the shared cataract phenotype observed in the affected siblings is consistent with the GCNT2 deficiency alone, an effect of this deletion on the function of TFAP2A and the observed phenotypes cannot be completely ruled out. Interestingly, a double second-cousin to the proband has been reported to be affected with bilateral anophthalmia/ microphthalmia (A/M), an ocular condition that is more consistent with the TFAP2A spectrum. It is possible that the familial deletion expanded to include TFAP2A in this patient; alternatively, the A/M diagnosis may have an independent genetic etiology. Unfortunately, no other familial samples were available for further study.
Conclusions
We identified a ~98-kb homozygous deletion involving several exons of GCNT2 and the region upstream of TFAP2A in two children affected with congenital cataracts from a consanguineous family of Pakistani decent. This cataract-causing deletion removes the first coding exons of GCNT2 isoforms A and B but leaves the GCNT2C sequence intact, providing further support for the isoform-specific roles of this gene; this is the first disruption of GCNT2 reported which does not affect isoform C. While the patients do not fit a diagnosis of BOFS, one sibling demonstrates mild overlap with the phenotypic spectrum, and therefore an effect of this deletion on the function of TFAP2A cannot be ruled out.
Abbreviations
- A/M:
-
Anophthalmia/Microphthalmia
- BOFS:
-
Branchio-ocular-facial-syndrome
- GCNT2:
-
Glucosaminyl (N-acetyl) transferase 2, I-branching enzyme
- TFAP2A:
-
Transcription factor AP-2 alpha
- WES:
-
Whole exome sequencing
References
Medsinge A, Nischal KK. Pediatric cataract: challenges and future directions. Clin Ophthalmol. 2015;9:77–90.
Shiels A, Hejtmancik JF. Genetics of human cataract. Clin Genet. 2013;84(2):120–7.
Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP, Costakos D, Yonath H, Hall S, Power P, Semina EV. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet. 2013;132(7):761–70.
Zhao L, Chen XJ, Zhu J, Xi YB, Yang X, Hu LD, Ouyang H, Patel SH, Jin X, Lin D, Wu F, Flagg K, Cai H, Li G, Cao G, Lin Y, Chen D, Wen C, Chung C, Wang Y, Qiu A, Yeh E, Wang W, Hu X, Grob S, Abagyan R, Su Z, Tjondro HC, Zhao XJ, Luo H, Hou R, Perry JJ, Gao W, Kozak I, Granet D, Li Y, Sun X, Wang J, Zhang L, Liu Y, Yan YB, Zhang K. Lanosterol reverses protein aggregation in cataracts. Nature. 2015;523(7562):607–11.
Chen P, Dai Y, Wu X, Wang Y, Sun S, Xiao J, Zhang Q, Guan L, Zhao X, Hao X, Wu R, Xie L. Mutations in the ABCA3 gene are associated with cataract-microcornea syndrome. Invest Ophthalmol Vis Sci. 2014;55(12):8031–43.
Berry V, Gregory-Evans C, Emmett W, Waseem N, Raby J, Prescott D, Moore AT, Bhattacharya SS. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet. 2013;21(12):1356–60.
Yu LC, Twu YC, Chang CY, Lin M. Molecular basis of the adult i phenotype and the gene responsible for the expression of the human blood group I antigen. Blood. 2001;98(13):3840–5.
Yu LC, Twu YC, Chou ML, Reid ME, Gray AR, Moulds JM, Chang CY, Lin M. The molecular genetics of the human I locus and molecular background explain the partial association of the adult i phenotype with congenital cataracts. Blood. 2003;101(6):2081–8.
Pras E, Raz J, Yahalom V, Frydman M, Garzozi HJ, Pras E, Hejtmancik JF. A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci. 2004;45(6):1940–5.
Lin M, Hou MJ, Yu LC. A novel IGnT allele responsible for the adult i phenotype. Transfusion. 2006;46(11):1982–7.
Borck G, Kakar N, Hoch J, Friedrich K, Freudenberg J, Nurnberg G, Yilmaz R, Daud S, Baloch DM, Nurnberg P, Oldenburg J, Ahmad J, Kubisch C. An Alu repeat-mediated genomic GCNT2 deletion underlies congenital cataracts and adult i blood group. Hum Genet. 2012;131(2):209–16.
Deml B, Reis LM, Lemyre E, Clark RD, Kariminejad A, Semina EV. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur J Hum Genet. 2016;24(4):535–41.
Krumm N, Sudmant PH, Ko A, O'Roak BJ, Malig M, Coe BP, NHLBI Exome Sequencing Project, Quinlan AR, Nickerson DA, Eichler EE. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–32.
D'haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50(4):262–70.
Schilter KF, Reis LM, Sorokina EA, Semina EV. Identification of an Alu-repeat-mediated deletion of OPTN upstream region in a patient with a complex ocular phenotype. Mol Genet Genomic Med. 2015;3(6):490–9.
Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67(3):183–93.
Volkmann BA, Zinkevich NS, Mustonen A, Schilter KF, Bosenko DV, Reis LM, Broeckel U, Link BA, Semina EV. Potential Novel Mechanism for Axenfeld-Rieger Syndrome: Deletion of a Distant Region Containing Regulatory Elements of PITX2. Invest Ophthalmol Vis Sci. 2011;52(3):1450–9.
Lauderdale JD, Wilensky JS, Oliver ER, Walton DS, Glaser T. 3' deletions cause aniridia by preventing PAX6 gene expression. Proc Natl Acad Sci U S A. 2000;97(25):13755–9.
Bhatia S, Kleinjan DA. Disruption of long-range gene regulation in human genetic disease: a kaleidoscope of general principles, diverse mechanisms and unique phenotypic consequences. Hum Genet. 2014;133(7):815–45.
Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, Burch MN, Clemens M, Mulliken JB, Smith R, Lin AE. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82(5):1171–7.
Dumitrescu AV, Milunsky JM, Longmuir SQ, Drack AV. A family with branchio-oculo-facial syndrome with primarily ocular involvement associated with mutation of the TFAP2A gene. Ophthalmic Genet. 2012;33(2):100–6.
Li H, Sheridan R, Williams T. Analysis of TFAP2A mutations in Branchio-Oculo-Facial Syndrome indicates functional complexity within the AP-2alpha DNA-binding domain. Hum Mol Genet. 2013;22(16):3195–206.
Acknowledgments
The authors also gratefully acknowledge the patients and their family for their participation in this research study. This work was supported by the National Institutes of Health awards R01EY015518 (EVS) and funds provided by the Children’s Hospital of Wisconsin (EVS), along with 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) program and the National Eye Institute of the National Institutes of Health under Award Number P30EY001931. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
This work was supported by the National Institutes of Health awards R01EY015518 (EVS) and funds provided by the Children’s Hospital of Wisconsin (EVS), along with 1UL1RR031973 from the Clinical and Translational Science Award (CTSA) program and the National Eye Institute of the National Institutes of Health under Award Number P30EY001931. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials
Data from this study that do not pertain to identifiable patient information are freely available and provided as supplemental material and/or can be obtained by contacting the corresponding author.
Authors’ contributions
EVS conceived and designed the study. DC performed ophthalmological evaluation and referred the patient to the study. LMR enrolled the family. HH and EW carried out the genetic studies. HH, EW, EVS, and LMR analyzed and interpreted the data. HH, EW, LMR, and EVS drafted the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent to publish
Written consent for publication was obtained from the parents of the patients. A copy of the consent is available for review by the Editor of this journal.
Ethics approval and consent to participate
This human study was approved by the Institutional Review Board of the Children’s Hospital of Wisconsin and carried out in accordance with the Declaration of Helsinki. Written and informed consent for molecular studies was obtained from the parents of the patients. A copy of the consent is available for review by the Editor of this journal.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Summary of PCR/qPCR reactions and copy number status in the affected family. (DOCX 21 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Happ, H., Weh, E., Costakos, D. et al. Case report of homozygous deletion involving the first coding exons of GCNT2 isoforms A and B and part of the upstream region of TFAP2A in congenital cataract. BMC Med Genet 17, 64 (2016). https://doi.org/10.1186/s12881-016-0316-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0316-0