
Abstract
Background
Macular corneal dystrophy (MCD) is a rare autosomal recessive disorder that is characterized by progressive corneal opacity that starts in early childhood and ultimately progresses to blindness in early adulthood. The aim of this study was to identify the cause of MCD in a black South African family with two affected sisters.
Methods
A multigenerational South African Sotho-speaking family with type I MCD was studied using whole exome sequencing. Variant filtering to identify the MCD-causal mutation included the disease inheritance pattern, variant minor allele frequency and potential functional impact.
Results
Ophthalmologic evaluation of the cases revealed a typical MCD phenotype and none of the other family members were affected. An average of 127 713 variants per individual was identified following exome sequencing and approximately 1.2 % were not present in any of the investigated public databases. Variant filtering identified a homozygous E71Q mutation in CHST6, a known MCD-causing gene encoding corneal N-acetyl glucosamine-6-O-sulfotransferase. This E71Q mutation results in a non-conservative amino acid change in a highly conserved functional domain of the human CHST6 that is essential for enzyme activity.
Conclusion
We identified a novel E71Q mutation in CHST6 as the MCD-causal mutation in a black South African family with type I MCD. This is the first description of MCD in a black Sub-Saharan African family and therefore contributes valuable insights into the genetic aetiology of this disease, while improving genetic counselling for this and potentially other MCD families.
Similar content being viewed by others


Background
Macular corneal dystrophy (MCD) (OMIM #217800) is a rare autosomal recessive disorder that is characterized clinically by irregularly shaped superficial opacities that progressively extend through the corneal stroma [1, 2]. Onset typically occurs in the first decade of life and progresses to severe bilateral visual impairment in adulthood, which ultimately necessitates keratoplasty [3]. MCD has been identified in a number of populations across the world [4–11], but it has not yet been reported in Sub-Saharan Africa.
Mutations in the carbohydrate sulfotransferase 6 gene (CHST6) were identified as the cause for MCD in 2000 [12]. CHST6 encodes corneal N-acetyl glucosamine-6-O-sulfotransferase (C-GlcNAc6ST), an enzyme which catalyses the sulfation of GlcNAc residues in the main glycosaminoglycan in the cornea, keratan sulfate, to generate sulfated keratan sulfate (KS). The defective sulfation of keratan sulfate that is caused by a deficiency in this enzyme leads to malformations in fibril organization in the cornea, which results in progressive corneal opacification in MCD patients [13–15].
MCD can be divided into two immunophenotypes based on the reactivity of the patient’s serum and corneal tissue to an antibody against KS. Antigenic KS reactivity is very low (or undetectable) in the serum and absent in corneal tissue in MCD type I patients, whereas normal to sub-normal KS levels are detectable in the serum and corneal stroma of type II MCD patients [16]. In situ hybridization analysis did not detect CHST6 transcripts in corneal epithelium of an MCD type II patient, suggesting that the mutations found in type II lead to a loss of cornea-specific expression of CHST6 [12]. These two subtypes are, however, clinically indistinguishable.
Patients with type I MCD usually harbour missense mutations and indels in the coding region of CHST6, which leads to the functional inactivation of the enzyme [12, 17], whereas deletions and/or rearrangements in the region between CHST6 and the neighbouring CHST5 have been reported in patients with MCD type II [1, 12]. This region likely harbours a gene regulatory element that affects the cell-specific transcription of CHST6 [12].
However, some studies failed to find potentially causative mutations in the coding region, upstream regulatory region or splice site mutations of CHST6 in MCD patients [1, 5, 6, 9, 18, 19]. Current theories to explain this include pathogenic mutations in an unknown regulatory promoter immediately upstream of CHST6 or cis-acting distant regulatory elements [1, 20], while genetic heterogeneity has not yet been excluded [1].
In the present study, which is the first description of MCD in a sub-Saharan African family, we used whole exome sequencing to identify the MCD-causal mutation in a consanguineous black South African family.
Methods
MCD Family
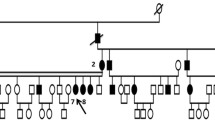
Two sisters presented with a complaint of decreased vision in 2007 to the Eye Clinic of the Charlotte Maxeke Johannesburg Academic Hospital in Johannesburg, South Africa. They were diagnosed with MCD based upon the distinctive clinical features, which were later confirmed by histopathologic examination following penetrating keratoplasty. We invited the entire family for an ophthalmic examination and seven additional family members consented to participate in this study. The pedigree was consistent with an autosomal recessive inheritance pattern and showed evidence of consanguinity (Fig. 1).

Schematic representation of the MCD pedigree. The 9 sequenced individuals are indicated with a + and their genotype for the E71Q mutation is indicated below the Individual ID. Individuals IV.3 and IV.4 (shaded black) show clinical manifestations of macular corneal dystrophy (MCD), while their consanguineous parents, siblings IV.1 and IV.6 and children did not
The study was approved by the Human Research Ethics Committee (Medical) of the University of the Witwatersrand, South Africa (protocol number M131125), and followed the tenets of the Declaration of Helsinki. Written informed consent was obtained from all participating family members for participation in this study and the use of their DNA and clinical data for research purposes after a genetic counsellor explained the nature and possible consequences of the study to them.
Complete ophthalmic examination of each participating family member was performed. Phenotyping included a slit-lamp examination, anterior segment photography and OCULUS Pentacam® examination.
Whole exome sequencing, assembly and variant calling
Targeted exome capture was performed by preparing sequencing libraries from genomic DNA using the NuGEN Ovation Ultralow DR Multiplex protocol followed by a SureSelectXT Human All Exon V5 + UTRs (70 MB) target enrichment (Agilent, Basel, Switzerland). Captured libraries were sequenced on the Illumina HiSeq 2500 with 76-bp paired-end reads. Reads were mapped to the human hg19 genome assembly using the Burrows-Wheeler Aligner (BWA) [21] and GATK base quality score recalibration, indel realignment, and duplicate removal was applied according to GATK Best Practises guidelines [22]. Variants were called with the GATK Unified Genotyper [23] and annotated with the Ensembl Variant Effect Predictor [24]. Variants with a VQSLOD score below 2 and/or a base coverage below 5x were considered low quality calls and consequently not included in the analysis.
Variant filtering to identify putative MCD-causal variant(s)
Using a tiered filtering strategy, we first explored variants that followed a simple autosomal recessive inheritance pattern (both affected sisters should be homozygous for the variant allele, carrier mother should be heterozygous and unaffected siblings and children can be either homozygous for the reference allele or heterozygous) (Fig. 2). We only considered novel variants and variants with a minor allele frequency (MAF) of less than 1 % in the 1000 Genomes Project (1kGP) [25] and NHLBI GO Exome Sequencing Project (ESP6500) databases, because developmental eye disorders, and MCD in particular, are considered very rare [26, 27].

Bioinformatic analysis pipeline
Non-coding or synonymous variants were excluded and the variants that remained following the filtering process were evaluated for possible functional impact using SIFT [28], PolyPhen-2 [29] and MutationTaster2 [30]. A database search of the genes containing potential MCD causal mutations was done using PubMed, Online Mendelian Inheritance in Man (OMIM) and the Human Gene Mutation Database (HGMD) Professional 2014.2 [31]. This filtering strategy was then repeated with compound heterozygous mutations that followed an autosomal recessive inheritance pattern in the family.
Validation and segregation analysis
Sanger sequencing was performed in each of the participating family members to confirm the segregation of the putative MCD-causal variant in the family. A fragment spanning the location of the CHST6 E71Q mutation was obtained by PCR amplification using sense 5′-AGGTCCAGATCCGTGGGTG-3′ and antisense 5′-CTTTCTGGTTTCCCGGCCA-3′ primers. These primers were then used to sequence the amplicons with the BigDye® Terminator v3.1 Cycle Sequencing chemistry (Thermo Fisher Scientific; Reinach, Switzerland). The purified fragment population was then loaded on a 3730XL Genetic Analyzer for the final capillary electrophoresis step.
Microscopy
Cross-sections of the corneal buttons excised during penetrating keratoplasty were stained with haematoxylin and eosin (HE). In addition, the following histochemical stains were performed: Alcian blue, Hale’s colloidal iron, Masson’s trichrome, Periodic acid-Schiff (PAS) and Congo red.
Results
Clinical evaluation
A South African Sotho-speaking family was identified with two MCD-affected sisters of consanguineous parents at the Eye Clinic of the Charlotte Maxeke Johannesburg Academic Hospital. Both affected sisters (IV.3 and IV.4; Fig. 1) presented to the clinic in September 2007 with the complaint of decreased vision. We recruited seven additional family members as shown in the multigenerational pedigree (Fig. 1).
Patient IV.3 was born in 1977. At the first examination her best-corrected visual acuity in both eyes was counting fingers (<20/200). Both corneas showed ill-defined opacities scattered throughout the stroma. Some were deep and peripheral and typical of macular dystrophy. The corneal stroma between the opacities also showed opacification. Pentacam and contact pachymetry were performed which revealed thin corneas. Corneal astigmatism was present measuring 4.2 dioptres in the right eye and 3.7 dioptres on the left. Intraocular pressures were normal and the eyes otherwise unremarkable, however the fundus view was poor.
Patient IV.4 was born in 1981. The first eye examination was similar to that of her sister, with a best-corrected visual acuity of counting fingers in either eye. Pentacam and contact pachymetry also revealed thin corneas. Corneal astigmatism was 2.4 dioptres on the right and 3.7 dioptres on the left. The intraocular pressures were normal, posterior polar cataracts were present bilaterally and view of the fundi was limited by the cloudy corneas.
Both patients subsequently had uneventful penetrating keratoplasties in their right eyes. The excised corneal tissue was preserved for microscopy and immunohistochemistry. At 3 months post-penetrating keratoplasty patient IV.3 had a clear graft with a best corrected visual acuity of 20/60 in the right eye. We are unsure about age of onset so amblyopia may be a factor in the end result. The lens was clear and fundus examination as well as posterior pole optical coherence tomography (OCT) was normal. Patient IV.4 had a clear graft with a best corrected visual acuity of 20/25 at 2 months post-penetrating keratoplasty in the right eye. Fundus examination and OCT of the posterior pole were unremarkable.
The following unaffected family members all underwent a comprehensive ophthalmological examination and were found to have no ocular pathology:
III.7, III.9, IV.1, IV.6, V.8, V.9, V.10, V.11.
Microscopy
Microscopic examination showed tissue compatible with derivation from the cornea (Fig. 3). Granular basophilic deposits were noted among the lamellae of the substantia propria, within the corneal endothelium, Descemet’s membrane and in Bowman’s membrane. The granular eosinophilic deposits were highlighted by Alcian blue, Hale’s colloidal iron and PAS stains. The granules were not seen on the Masson trichrome stain. The Congo red stain was negative. No birefringent material was seen on polarising microscopy.

Clinical phenotypes. a. Pre-operative photograph of the right eye of IV.3 demonstrating ill-defined corneal stromal opacities. b. Cornea of IV.3. Hale’s colloidal iron 20x. The presence of the granular deposits (arrows) are highlighted by a Hale’s colloidal iron stain. c. Cornea of IV.3. Hale’s colloidal iron 100x. Higher magnification demonstrates the presence of the granular deposits (arrows) among the lamellae of the substantia propria and within the corneal endothelium. d. Control cornea: Hale’s colloidal iron 40x. Control cornea stained with Hale’s colloidal iron fails to identify the presence of basophilic deposits within the substantia propria
Whole exome sequencing and variant filtering
We performed whole exome sequencing on 9 family members to an average depth of 43x. On average, 98 and 95 % of bases were covered to 5x and 10x within the targeted regions, respectively. We identified an average of 127 713 small variants (<50 bp) per individual after filtering out the low quality base calls and variants with a coverage below 5x in the whole family. Approximately 1.2 % of these variants were not present in any of the investigated public databases (Table 1).
The two affected sisters shared 106 552 variants that varied from the human reference genome in either a heterozygous or homozygous state (Fig. 2). This number was reduced to 1 855 variants when we filtered based on a simple autosomal recessive mode of inheritance by only keeping the variants where the affected sisters are homozygous for the variant allele, carrier mother is heterozygous and the unaffected siblings and children are either homozygous for the reference allele or heterozygous. We then retained the 81 variants with a MAF below 1 % in the 1kGP and ESP6500 data sets. Table 2 shows the 7 variants that remained after we filtered out variants with a low likelihood of adverse functionality based on the type of mutation as assessed using SIFT, PolyPhen-2 and MutationTaster2.
Following the same approach we identified compound heterozygous variants in 9 genes, which followed the autosomal recessive inheritance pattern and where both mutations survived our frequency and functionality filters (Additional file 1: Table S1).
Putative MCD-causal mutation
The most probable MCD-causal mutation identified is a E71Q (c.211G > C) mutation in the CHST6 gene, which is a known MCD-causal gene. Sanger sequencing confirmed that this mutation segregated with the disease in the family (Fig. 4) and it was absent from the 1kGP, ESP6500, dbSNP, ClinVar, HGMD and African Genome Variation Project (AGVP) [32] data sets.

Results form CHST6 Sanger Sequencing. Electropherograms from Sanger sequencing indicating homozygous E71Q genotype in the two affected sisters (IV.3 and IV.4), heterozygous genotype in the carrier mother (III.8) and homozygous reference genotype in the unaffected brother (IV.1). The red arrow indicates the position of the E71Q mutation and the sequences are given for the reverse strand
This E71Q mutation results in a non-conservative amino acid change as it replaces a negatively charged amino acid (glutamic acid) with an amino acid with polar uncharged side chains (glutamine). All mutations in CHST6 that alter a codon are presumed to be of pathogenic significance due to the fact that this gene contains large blocks of sequences that are predicted to form a tertiary structural domain that is conserved among sulfotransferase enzymes [1, 5, 18, 33]. The E71 amino acid is conserved in all but two of the human carbohydrate sulfotransferases [5] and falls within the human CHST6 functional domain (Sulfotransfer_3; ID:PF13469) [34].
Variants of uncertain significance
We identified potentially damaging homozygous mutations of unknown clinical relevance in RP1L1 and CSMD1, which also segregated with MCD in the family (Table 2). None of the identified compound heterozygous mutations are believed to be of any pathogenic relevance based on gene function, their MAF in the general population and bioinformatic predictors of functional impact (Additional file 1: Table S1).
RP1L1 is associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy [35, 36]. The S564C mutation identified here is recorded in dbSNP131 (rs77585543) and is predicted to be damaging according to SIFT and PolyPhen-2, but not MutationTaster2. It has a MAF of 4 % in the African sample subset of the 1kGP and two of these individuals are homozygous for this mutation.
We confirmed that RP1L1 is expressed in retinal cells from healthy donors and found that RP1L1 expression levels are very low in the cornea, although higher levels of RP1L1 are detected in the cornea than in negative control samples (human dermal fibroblasts) (Additional file 1: Figure S1). Taken together, these data suggest that the RP1L1 mutation is unlikely to be the MCD-causal mutation in this family. It is perhaps plausible that this mutation might somehow impact on the retinal phenotype of these patients, but we did not observe any unusual retinal features.
A g.1501249C > G splice site mutation (rs368653091) in CSMD1 that segregates with MCD in the family is predicted to be disease causing according to MutationTaster2 as it changes the sequence motif adjacent to the acceptor sequence at position 1501247 (Table 2). CSMD1 encodes the CUB and Sushi multiple domains 1 protein, which is associated with phenotypic variance in neuropsychological disorders [37–39], but it has no documented effect on ocular phenotypes. It is therefore unlikely that this splice site mutation will drive the MCD phenotype in the affected sisters.
Discussion
We report on a consanguineous black South African family with type I MCD, which is caused by a novel E71Q mutation in CHST6. It is the first report of MCD in a Sub-Saharan African family, although it is not the first report of MCD in an individual with African ancestry as Patel et al. previously identified a novel mutation in CHST6, which is associated with MCD type II in an African American [40].
The CHST6 gene encodes the C-GlcNAc6ST enzyme, which is responsible for generating KS in the cornea. Improper functioning of this enzyme leads to malformations in fibril organization in the cornea, which manifests as the characteristic superficial opacities seen in MCD patients [13–15]. However, some studies failed to identify mutations in the CHST6/CHST5 region in MCD patients [1, 5, 6, 9, 18, 19]. We used a whole exome sequencing approach to investigate the cause of MCD in this South African family, because we did not have sufficient African data to inform on whether a candidate gene approach would be successful in this case.
We identified a homozygous E71Q mutation in the two affected sisters that falls within the CHST6 functional domain, which segregated with MCD in the family. El-Ashry et al. previously identified a G905T transversion that results in another amino acid change at the same position (E71D) in a British family with MCD. However, this mutation was not considered deleterious as it leads to a conservative amino acid change and was inherited in tandem with a P72S non-conservative amino acid substitution and the authors concluded that P72S, rather than the E71D mutation, was the causal mutation in this family [41].
The E71Q mutation identified here results in a change from a negatively charged glutamic acid to the polar uncharged glutamine at a residue that is highly conserved among human sulfotransferases [1, 5]. It is, furthermore, not present in the 1kGP, ESP6500, dbSNP, ClinVar or HGMD databases and three programs for analysing protein functions, Polyphen2, SIFT and MutationTaster2, predicted that the E71Q mutation is probably damaging, deleterious and disease causing, respectively. Taken together, this suggests a very high likelihood that this mutation would have a deleterious effect on the C-GlcNAc6ST enzyme.
We identified two additional variants of unknown clinical relevance that segregated with MCD in our family: a g.1501249C > G splice site mutation in CSMD1 and a S564C missense muation in RP1L1. The CSMD1 mutation is predicted to be disease causing by MutationTaster2. Aberrant splicing in CSMD1 has been associated with cancer susceptibility [42], while some studies report association between CSMD1 mutations and phenotypic variance in neuropsychological disorders [37–39], but it has no documented effect on any ocular phenotype. In silico predictions of functionality of splicing variants are, furthermore, more vulnerable to false positive predictions than similar predictors for missense variants [43]. The CSMD1 splice variant identified here is therefore not the most likely driver of the MCD phenotype in this family.
It is possible to speculate that the RP1L1 S564C mutation might somehow have an effect on the clinical phenotype in these MCD patients due to RP1L1’s association with two retinal disorders, occult macular dystrophy and retinitis pigmentosa. However, we found that this gene is not expressed in the human cornea, making it unlikely to have an impact on the corneal phenotype of this family. We did confirm that this gene is expressed in the human retina, but fundus examination of both individuals post penetrating keratoplasty was normal and the post-operative visual acuities were appropriate for the degree of astigmatism and did not suggest retinal disease. The S564C mutation is also observed twice in a homozygous state in the 1kGP data. Taken together, this suggests that the S564C mutation is unlikely to cause a severe ocular phenotype in this family.
A large number of in silico functional predictors are available at present, all of which aim to address the difficult task of prioritizing variants in disease gene identification studies based on different types of data, often yielding different results [44]. However, all of these predictors report an error rate and the studies that aim to evaluate these predictors are slightly flawed due to the overlap in the training and test data sets, which may yield overly optimistic results [45]. It is therefore important to note that in silico functional predictors serve as a context within which to interpret sequencing results and that multiple lines of evidence are needed to infer pathogenicity.
African populations contributed the largest number of variants and with the highest fraction of novel variants in the 1kGP [25]. This reflects the great genetic diversity in populations with African ancestry, which has been demonstrated in a number of additional studies [46, 47]. We identified an average of 1569 novel variants per individual. An average of 11 100 of the identified variants were non-synonymous coding (missense) mutations and almost a third of these variants are novel, which is higher than the average recorded in the 1kGP [25].
It is often difficult to get an early, accurate diagnosis for patients with rare diseases, particularly in an overburdened public health care system. The identification of the MCD causal mutation is of great significance to this family as it provides valuable information on the risks to other family members and improves accurate genetic counselling. Fortunately all members of this family that were tested could be reassured that they did not have the same disorder as the two affected sisters. This was especially relevant to the younger family members. Tools for early diagnosis equip the family with the knowledge to seek timely, effective care for other family members with vision problems. Furthermore, our study contributes research into the genetic underpinnings of ocular disease phenotypes in African populations. Studies on African populations have a high probability of identifying novel disease genes due to their high degree of genetic diversity. Such studies additionally serve as a guide to determine where data generated on Caucasian populations might be transferable to Africans.
Conclusions
We have identified a novel E71Q mutation in CHST6 as the cause of MCD in this black South African family. This is the first clinical description of MCD in a Sub-Saharan African family as well as the first investigation into the genetic aetiology of MCD in a Sub-Saharan African population. This study therefore contributes towards genetic counselling for MCD patients of African descent.
References
Klintworth GK, Smith CF, Bowling BL. CHST6 mutations in North American subjects with macular corneal dystrophy : a comprehensive molecular genetic review. Mol Vis. 2006;12:159–76.
Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7.
Sari ES, Kubaloglu A, Unal M, Pinero D, Bulut N, Erol MK, et al. Deep anterior lamellar keratoplasty versus penetrating keratoplasty for macular corneal dystrophy: a randomized trial. Am J Ophthalmol. 2013;156:267–74.e1.
Lee YK, Chang D-J, Chung SK. A case of Korean patient with macular corneal dystrophy associated with novel mutation in the CHST6 gene. Korean J Ophthalmol. 2013;27:454–8.
Aldave AJ, Yellore VS, Thonar EJ, Udar N, Warren JF, Yoon MK, et al. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am J Ophthalmol. 2004;137:465–73.
Niel F, Ellies P, Dighiero P, Soria J, Sabbagh C, San C, et al. Truncating mutations in the carbohydrate sulfotransferase 6 gene (CHST6) result in macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:2949–53.
El-Ashry MF, Abd El-Aziz MM, Shalaby O, Bhattacharya SS. Molecular genetic study of Egyptian patients with macular corneal dystrophy. Br J Ophthalmol. 2010;94:250–5.
Ha NT, Chau HM, Cung LX, Thanh TK, Fujiki K, Murakami A, et al. Mutation analysis of the carbohydrate sulfotransferase gene in Vietnamese with macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2003;44:3310–6.
Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, et al. Molecular genetic analysis of macular corneal dystrophy patients from North India. Ophthalmic Res. 2012;48:28–32.
Klintworth GK, Oshima E, Al-Rajhi A, Al-Saif A, Thonar EJ, Karcioglu ZA. Macular corneal dystrophy in Saudi Arabia: a study of 56 cases and recognition of a new immunophenotype. Am J Ophthalmol. 1997;124:9–18.
Liu N-P, Smith CF, Bowling BL, Jonasson F, Klintworth GK. Macular corneal dystrophy types I and II are caused by distinct mutations in the CHST6 gene in Iceland. Mol Vis. 2006;12:1148–52.
Akama TO, Nishida K, Nakayama J, Watanabe H, Ozaki K, Dota A, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet. 2000;26:237–41.
Pomin VH. Keratan sulfate: an up-to-date review. Int J Biol Macromol. 2015;72:282–9.
Musselmann K, Hassell JR. Focus on molecules: CHST6 (carbohydrate sulfotransferase 6; corneal N-acetylglucosamine-6-sulfotransferase). Exp Eye Res. 2006;83:707–8.
Quantock AJ, Young RD, Akama TO. Structural and biochemical aspects of keratan sulphate in the cornea. Cell Mol Life Sci. 2010;67:891–906.
Saito T, Nishida K, Nakayama J, Akama TO, Fukuda MN, Watanabe K, et al. Sulfation patterns of keratan sulfate in different macular corneal dystrophy immunophenotypes using three different probes. Br J Ophthalmol. 2008;92:1434–6.
Akama TO, Nakayama J, Nishida K, Hiraoka N, Suzuki M, McAuliffe J, et al. Human corneal GlcNac 6-O-sulfotransferase and mouse intestinal GlcNac 6-O-sulfotransferase both produce keratan sulfate. J Biol Chem. 2001;276:16271–8.
Sultana A, Sridhar MS, Klintworth GK, Balasubramanian D, Kannabiran C. Allelic heterogeneity of the carbohydrate sulfotransferase-6 gene in patients with macular corneal dystrophy. Clin Genet. 2005;68:454–60.
Birgani SA, Salehi Z, Houshmand M, Mohamadi MJ, Promehr LA, Mozafarzadeh Z. Novel mutations of CHST6 in Iranian patients with macular corneal dystrophy. Mol Vis. 2009;15:373–7.
Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73.
Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A. Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2011;52:6959–63.
Bermejo E, Martínez-Frías ML. Congenital eye malformations: clinical-epidemiological analysis of 1,124,654 consecutive births in Spain. Am J Med Genet. 1998;75:497–504.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–82.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods NIH Public Access. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9.
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–32.
Kakuta Y, Pedersen LG, Pedersen LC, Negishi M. Conserved structural motifs in the sulfotransferase family. Trends Biochem Sci. 1998;23:129–30.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30.
Akahori M, Tsunoda K, Miyake Y, Fukuda Y, Ishiura H, Tsuji S, et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am J Hum Genet. 2010;87:424–9.
Davidson AE, Sergouniotis PI, Mackay DS, Wright GA, Waseem NH, Michaelides M, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat. 2013;34:506–14.
Lotan A, Fenckova M, Bralten J, Alttoa A, Dixson L, Williams RW, et al. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front Neurosci. 2014;8:331.
Xu W, Cohen-Woods S, Chen Q, Noor A, Knight J, Hosang G, et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet. 2014;15:2.
Steen VM, Nepal C, Ersland KM, Holdhus R, Nævdal M, Ratvik SM, et al. Neuropsychological deficits in mice depleted of the schizophrenia susceptibility gene CSMD1. PLoS One. 2013;8:e79501.
Patel DA, Harocopos GJ, Chang S-H, Vora SC, Lubniewski AJ, Huang AJ. Novel CHST6 gene mutations in 2 unrelated cases of macular corneal dystrophy. Cornea. 2011;30:664–9.
El-Ashry MF, Abd El-Aziz MM, Wilkins S, Cheetham ME, Wilkie SE, Hardcastle AJ, et al. Identification of novel mutations in the carbohydrate sulfotransferase gene (CHST6) causing macular corneal dystrophy. Invest Ophthalmol Vis Sci. 2002;43:377–82.
Ma C, Quesnelle KM, Sparano A, Rao S, Park MS, Cohen MA, et al. Characterization CSMD1 in a large set of primary lung, head and neck, breast and skin cancer tissues. Cancer Biol Ther. 2009;8:907–16.
Houdayer C. In silico prediction of splice-affecting nucleotide variants. Methods Mol Biol. 2011;760:269–81.
Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: state-of-the-art web-based tools. Brief Bioinform. 2013;14:448–59.
Grimm DG, Azencott C-A, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015;36:n/a – n/a.
Campbell MC, Tishkoff SA. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet. 2008;9:403–33.
Campbell MC, Tishkoff SA. The Evolution of Human Genetic and Phenotypic Variation in Africa Review. Curr Biol Elsevier Ltd. 2010;20:R166–73.
Acknowledgements
We would like to thank the family for their participation and cooperation. Research reported in this publication was supported by the Fogarty International Center of the National Institutes of Health under Award Number D43 TW008330. MR is a South African Research Chair in Genomics and Bioinformatics of African populations hosted by the University of the Witwatersrand, funded by the Department of Science and Technology and administered by National Research Foundation of South Africa (NRF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Fogarty International Centre, or the National Institutes of Health or the NRF. Genetic sequencing and analysis was performed during NC’s participation in the Novartis and University of Basel Next Generation Scientist program, a 3-month research internship in Basel, Switzerland that aims to build scientific and leadership capability in emerging country scientists. In particular, we would like to thank Moritz Frei for the library preparation and exome sequencing; Anita Fernandez, Mariavittoria Iazeolla and Simone Popp for their technical assistance and Fan Yang and Bolan Linghu for their support and assistance with data analysis during this time. This research was further supported by a self-initiated research grant from the South African Medical Research Council (TC).
Availability of data and materials
Information relative to the interpretation of the data is available in Supplementary information. The exome sequencing data files are available on request from #Rsch-SBIMB@wits.ac.za or from the corresponding author.
Authors’ contributions
NC, SW, SG, TC and MR wrote the manuscript. SW, SG and TC recruited and evaluated the family and assisted with the clinical interpretation of results. NC, MSC and CZ carried out the bioinformatic analysis. SB-M, MS, FS, NC, MR, DSR, AL, KP and PS conceived and designed experiments and assisted with the interpretation of results. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from all participating family members (or parents in the case of minors) for publication of this research and any accompanying clinical data and images after a genetic counsellor explained the nature and possible consequences of the study to them. A copy of the written consent is available for review by the Editor of this journal.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Results from variant filtering for compound heterozygous that segregated with MCD in the family with an autosomal recessive inheritance pattern. Figure S1. Results form qRT-PCR investigating RP1L1 expression levels in the cornea. (DOCX 121 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carstens, N., Williams, S., Goolam, S. et al. Novel mutation in the CHST6 gene causes macular corneal dystrophy in a black South African family. BMC Med Genet 17, 47 (2016). https://doi.org/10.1186/s12881-016-0308-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0308-0