
Abstract
Background
Ring chromosome 18 [r(18)] is formed by 18p- and 18q- partial deletion and generates a ring chromosome. Loss of critical genes on each arm of chromosome 18 may contribute to the specific phenotype, and the clinical spectrum varieties may heavily depend on the extent of the genomic deletion. The aim of this study is to identify the detailed breakpoints location and the deleted genes result from the r18.
Case presentation
Here we describe a detailed diagnosis of a seven-year-old Chinese girl with a ring chromosome 18 mutation by a high-throughput whole-genome low-coverage sequencing approach without karyotyping and other cytogenetic analysis. This method revealed two fragment heterozygous deletions of 18p and 18q, and further localized the detailed breakpoint sites and fusion, as well as the deleted genes.
Conclusions
To our knowledge, this is the first report of a ring chromosome 18 patient in China analyzed by whole-genome low-coverage sequencing approach. Detailed breakpoints location and deleted genes identification help to estimate the risk of the disease in the future. The data and analysis here demonstrated the feasibility of next-generation sequencing technologies for chromosome structure variation including ring chromosome in an efficient and cost effective way.
Similar content being viewed by others


Background
Ring chromosome 18 [r(18)] is formed from breakage of both ends of the chromosome and the break ends generate a ring chromosome [1]. Individuals with r(18) have 18p and 18q partial deletions and according phenotype, such as microcephaly, mental deficiency, hypotonia, and congenital heart defects [2, 3]. Short stature, microcephaly, mental deficiency, craniofacial dysmorphism and extremity abnormalities are the most commonly reported features in patients with r(18). The phenotype with r(18) syndrome is highly variable and depends on the combination of 18p- syndrome and 18q- syndrome. Loss of critical genes on each arm of chromosome 18 may contribute to the specific symptoms, and the clinical spectrum varieties may heavily depend on the extent of the genomic deletion [4].
Whole-genome low-coverage sequencing has been reported previously by our group to accurately detect chromosomal structural variation-associated breakpoints and affected region without cytogenetic analysis on patients [5].
In the current study, we applied whole-genome low-coverage sequencing to characterize the ring chromosome 18 mutation at a molecular level in a Chinese young girl for the first time. We described the full profile of clinical examination, genetic characterizations, and clinical treatment report. We localized the genomic breakpoints as well as identified the deleted genes. The deletion of the genes and detailed breakpoint identified help to understand the genotype- correlation and estimate the risk of the disease in the future.
Case presentation
The patient was born to non-consanguineous at the year of 2006. The patient was born at 40 weeks gestation with a birth weight of 3,050 g and length of 49 cm. At 2 years of age, she was found shorter than children of the same age. In April 2008, she was diagnosed hypothyroidism in the local clinic. Replacement of thyroid hormone (levothyroxine) was started for the treatment of autoimmune hypothyroidism. Unregular treatment lasted one year and discontinue by parents themselves.
At 6 and half years of age (March,2013), she came to our hospital for short stature. At the time of our first evaluation, she had a short stature problem (height: 90.7 cm [−6.0SD, equivalently 50 percentile of 2–2.5 years old], weight: 12.0 kg [<3 percentile, equivalently 50 percentile of 2–2.5 years old]). The general examination phenotypes of this patient include intellectual disability with IQ = 70, hypoactive, poor appetite, hypotonia, short neck without webbing, short fingers and toes, much shorter fifth finger, sparse hair and dry skin. She had dry stool once every 1 ~ 3 days. No goiter, lymphadenopathy or hepatosplenomegaly were noted.
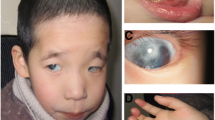
The facial appearance of the patient was including flat midface, puffy eyelids, hypertelorism, epicanthic fold, flat nasal bridge, and micrognathia. Wide mouth, downturned corners of mouth, thick lips, large protruding ears ptosis and upslanting palpebral ptosis were also noted (Fig. 1). High narrow palate and several cavities in teeth were observed. In addition, she suffered from bronchitis and otitis media frequently, without serious infections. Auscultation revealed no heart murmur and normal respiratory sounds.

Abnormalities of the craniofacial appearance. Facial appearance of the patient at age 7, showing flat midface, puffy eyelids, hypertelorism, epicanthic fold, flat nasal bridge, and micrognathia. a frontal view. b lateral view
Serological examination results showed normal liver and kidney functions but abnormal thyroid function, which prompted central autoimmune hypothyroidism and autoimmune thyroiditis. The thyroid auto antibodies were positive. Both TPO-Ab and TG-Ab were extremely high. The levels of IGF-1 and IGF-BP3 decreased drastically. IgA was slightly increased. E2、PROG、PRL and TESTO were all normal (data not show). Flow cytometry detection of T cell subgroup revealed that CD3 and CD8 + T were slightly higher (Additional file 1: Table S1). After euthyrox therapy, her total cholesterol and the triglyceride were back to normal levels, but the lipoprotein-α was still high (494.2 mg/l, reference range: 0-300 mg/l), the IGF-1 still low (29.8 ng/ml, reference range:64-345 ng/ml).
The abdominal color ultrasound results showed normal liver, uterus and ovaries. The sizes of both kidneys were smaller than normal. (left kidney: 6.2 cm × 2.8 cm, right kidney: 5.7 cm × 2.4 cm). The thyroid color ultrasound revealed that the thyroid was enlarged and its echo was not uniform. The thyroid isthmus was 0.5 cm thick (left lobe thyroid: 2.9 cm × 1.0 cm × 1.2 cm, right lobe thyroid: 3.3 cm × 1.0 cm × 1.3 cm) accompanied with uneven internal spots and echoes, like a network. The cardiac color ultrasound showed that the structure, shape and valves of the heart had no obvious abnormality.
The MRI image results revealed that the Pituitary height was 1.0 mm, much smaller than the normal size, and the neurohypophysis was not seen clearly, which indicated pituitary dysplasia (Fig. 2).

Pituitary gland on MRI. The MRI image results revealed that the Pituitary height was 1.0 mm, much smaller than the normal size, and the neurohypophysis was not seen clearly, which indicated pituitary dysplasia. a Coronal MRI scan of Pituitary Gland. b Sagittal MRI scan of Pituitary Gland
Following informed consent, hromosomal analysis was performed on peripheral blood lymphocyte cultures. The result of conventional karyotyping was 46, XX, r (18) (Fig. 3). No chromosomal anomaly was detected in either of parent by karyotyping analysis (data not shown).

Patient’s Karyotype analysis result by G-banding technique. The result of conventional karyotyping showed 46, XX, r(18) (arrow indicated)
Patient’s genomic DNA was extracted from peripheral blood using Qiagen DNA extraction kit and then was used to construct DNA libraries and to do sequencing assay including base calling. After removing reads containing sequencing adaptors and low quality reads, the high quality pair-end reads were aligned to the NCBI human reference genome (hg19, GRCh37.1) using SOAP2 [6]. Only uniquely mapped reads were remained in the following analysis.
The ring chromosome variation could be discovered using chimeric read pairs, which are paired-end reads that mapped to two different chromosomes.. The detail steps are listed in our previous published study [5].
Finally, we identified two partial deletions which are a portion of 18p from 1 bp to nearly 3,881,000 bp (3.88 Mb), and a portion of 18q from nearly 73,239,191 bp to terminal (4.83 Mb) base on the bioinformatics results. Both ends of chromosome 18 showed heterozygous terminal deficiency (Fig. 4). The remaining sequence of chromosome 18 generated a ring from breakage and subsequent fusion of both chromosome arms. The two breakpoints located in 18p11.31 band and 18q23 band respectively. The detailed breakpoint sites were validated to be at 3,880,565 bp and at 73,239,237 bp of chr18 respectively by Sanger sequencing. Besides, we also found a 20 bp insertion between the fusion breakpoints (Fig. 5). There were 19 genes deleted at chromosome 18 (pter → p11.31) and 12 genes deleted at chromosome 18 (q23 → qter) (Tables 1 and 2).

The Copy Number Ratio of Chromosome 18. The blue line indicates the normal diploid copy number ratio, and the window size is 5 kb. Both ends of chromosome 18 has a partial deletion, the copy number ratio is 0.5 showed heterozygous terminal deficiency. Centromere starts from 150Kb to 180Kb

The Sanger sequence alignment around the breakpoints of chromosome 18. We found there are a 20 bp insertion between the fusion breakpoints
Discussion
Ring chromosome 18 syndrome is a rare human cytogenetic abnormality. The syndrome is formed from breakage of both ends of the chromosome and the break ends generate a ring chromosome. The phenotype with r(18) syndrome is highly variable and depends on the combination of 18p- syndrome and 18q- syndrome [7]. The deletion of the short arm of chromosome 18 became a well-known chromosomal aberration after first discovery by de Grouchy in 1963 [2]. In 2009, Patricia et al. analyzed 18q in a high resolution level using aCGH, although they clarified the detailed breakpoint location, the deleted genes result from breakage of 18q were not able to be identified [8]. Normally, people use conventional karyotyping, FISH or aCGH to analysis chromosome aberrations, however, these methods have their limitations of revealing responsible critical genes and clarifying the genotype-phenotype correlations. Whole-genome low-coverage sequencing analysis could solve these problems at a base-level resolution.
Immunoglobulin A deficiency is frequently associated with ring chromosome 18 syndrome [9]. However, IgA deficiency was not noted in our patient, and further our patient appears features of central autoimmune hypothyroidism and small pituitary glands. The pituitary glands of our patient appeared morphologically small on head magnetic resonance imaging, while the thyroid showed morphologically normal on ultrasound. After receiving 10 months hormone therapy (levothyroxine), the IGF-1, T3 and T4 levels were still low, indicating that small pituitary invoked some functional defects, which resulted in the negative feedback failure of Hypothalamus-hypophysis-thyroid axis (HHTA).
There were totally 31 genes deleted at the del(18p) and del(18q) region. Some of them are very important for the physiological activity of the cells. Such as the USP14 gene that encodes a member of the ubiquitin-specific processing (UBP) family of proteases that is a deubiquitinating enzyme (DUB). Mice with a mutation that results in reduced expression of the ortholog of this protein are retarded for growth [10]. Gripp et al. [11] concluded that TGIF1 links the NODAL signaling pathway to the bifurcation of the human forebrain and the establishment of ventral midline structures. The GALR1 gene is widely expressed in the brain and spinal cord, as well as in peripheral sites such as the small intestine and heart [12]. Mutations in CTDP1 gene are associated with congenital cataracts, facial dysmorphism and neuropathy syndrome (CCFDN) [13]. So that, the inactivity of these genes may results to neurodevelopment, craniofacial appearance, oral manifestations and brain development anomalies.
In this report, We have presented a ring chromosome 18 patient with two heterozygous deletions of 3.88 Mb and 4.83 Mb indentified by whole-genome low-coverage sequencing method. The deletion of the genes and ring closure of chromosome 18 contribute to the clinical picture of dysmorphogenesis and mental retardation. Detailed breakpoints location and deleted genes identification help to estimate the risk of the disease in the future. At the same time, further studies are needed to delineate the function of responsible critical genes and clarify the genotype-phenotype correlations. The report here demonstrated the feasibility of next-generation sequencing technologies for chromosomal structural variation including ring chromosome in an efficient and cost effective way, which would improve the detection and prediction of genotype and phenotypic outcomes to direct postnatal medical care.
Conclusions
In conclusion, we analyzed a ring chromosome 18 patient in China by whole-genome low-coverage sequencing method for the first time. We described the full profile of clinical examination, genetic characterizations, and clinical treatment report. We localized the genomic breakpoints as well as identified the deleted genes. Detailed breakpoints location and deleted genes identification help to estimate the risk of the disease in the future. The report here demonstrated the feasibility of next-generation sequencing technologies for chromosomal structural variation including ring chromosome in an efficient and cost effective way, which would improve the detection and prediction of genotype and phenotypic outcomes to direct postnatal medical care.
Consent
Written informed consent was obtained in accordance with the Institutional Review Board of Wuhan Maternal and Child Health Hospital and the Declaration of Helsinki. The parents permitted the publication of the case, their clinical details and images.
References
Wertelecki W, Gerald PS. Clinical and chromosomal studies of the 18q- syndrome. J Pediatr. 1971;78(1):44–52.
De Grouchy J, Leveque B, Debauchez C, Salmon C, Lamy M, Marie J. [17–18 Ring-Chromosomes and Congenital Malformations in a Young Girl]. Ann Genet. 1964;7:17–23.
Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, Hoffman DS, Muller S, Schaub RL, Leach RJ. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet. 1999;85(5):455–62.
Subrt I, Pokorny J. Familial occurrence of 18q. Humangenetik. 1970;10(2):181–7.
Dong Z, Jiang L, Yang C, Hu H, Wang X, Chen H, Choy KW, Hu H, Dong Y, Hu B. A robust approach for blind detection of balanced chromosomal rearrangements with whole-genome low-coverage sequencing. Hum Mutat. 2014;35(5):625–36.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7.
Brkanac Z, Cody JD, Leach RJ, DuPont BR. Identification of cryptic rearrangements in patients with 18q- deletion syndrome. Am J Hum Genet. 1998;62(6):1500–6.
Heard PL, Carter EM, Crandall AC, Sebold C, Hale DE, Cody JD. High resolution genomic analysis of 18q- using oligo-microarray comparative genomic hybridization (aCGH). Am J Med Genet A. 2009;149A(7):1431–7.
Litzman J, Brysova V, Gaillyova R, Thon V, Pijackova A, Michalova K, Zemanova Z, Lokaj J. Agammaglobulinaemia in a girl with a mosaic of ring 18 chromosome. J Paediatr Child Health. 1998;34(1):92–4.
Wilson SM, Bhattacharyya B, Rachel RA, Coppola V, Tessarollo L, Householder DB, Fletcher CF, Miller RJ, Copeland NG, Jenkins NA. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet. 2002;32(3):420–5.
Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25(2):205–8.
Walli R, Schafer H, Morys-Wortmann C, Paetzold G, Nustede R, Schmidt WE. Identification and biochemical characterization of the human brain galanin receptor. J Mol Endocrinol. 1994;13(3):347–56.
Varon R, Gooding R, Steglich C, Marns L, Tang H, Angelicheva D, Yong KK, Ambrugger P, Reinhold A, Morar B. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet. 2003;35(2):185–9.
Acknowledgments
We thank all participants involved in this study.
Authors’ contributions
HY, CY and YY managed the project. XH, LY, YQ and PT. collected and prepared the samples. FM and DY performed the sequencing. CY and WZ performed the bioinformatics analysis. HY and CY wrote the paper. LL, DY and YY revised the paper.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
The serological examination results. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yao, H., Yang, C., Huang, X. et al. Breakpoints and deleted genes identification of ring chromosome 18 in a Chinese girl by whole-genome low-coverage sequencing: a case report study. BMC Med Genet 17, 49 (2016). https://doi.org/10.1186/s12881-016-0307-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0307-1