
Abstract
Background
Waardenburg syndrome type I (WS1), an auditory-pigmentary genetic disorder, is caused by heterozygous loss-of-function mutations in PAX3. Abnormal physical signs such as dystopia canthorum, patchy hypopigmentation and sensorineural hearing loss are common, but short stature is not associated with WS1.
Case presentation
We reported a 4-year and 6 month-old boy with a rare combination of WS1 and severe short stature (83.5 cm (−5.8SD)). His facial features include dystopia canthorum, mild synophrys, slightly up-slanted palpebral fissure, posteriorly rotated ears, alae nasi hypoplasia and micrognathia. No heterochromia was noticed. He had a normal intelligence quotient and hearing. Insulin-like growth factor-1 (IGF-1) was 52.7 ng/ml, lower than the normal range (55 ~ 452 ng/ml) and the peak growth hormone level was 7.57 ng/ml at 90 minutes after taking moderate levodopa and pyridostigmine bromide. The patient exhibited a good response to human growth hormone (rhGH) replacement therapy, showing a 9.2 cm/year growth rate and an improvement of 1 standard deviation (SD) of height after one year treatment. CMA test of patient’s DNA revealed a 4.46 Mb de novo deletion at 2q35-q36.2 (hg19; chr2:221,234,146-225,697,363).
Conclusions
PAX3 haploinsufficiency is known to cause Waardenburg syndrome. Examining overlapping deletions in patients led to the conclusion that EPHA4 is a novel short stature gene. The finding is supported by the splotch-retarded and epha4 knockout mouse models which both showed growth retardation. We believe this rare condition is caused by the haploinsufficiency of both PAX3 and EPH4 genes. We further reported a growth response to recombinant human growth hormone treatment in this patient.
Similar content being viewed by others

Background
Waardenburg syndrome (WS) is a group of auditory-pigmentary genetic disorders caused by neural crest development defects. Mutations in PAX3, a transcription factor belonging to the family of paired-box-containing proteins, causes WS type 1 (WS1, OMIM # 193500) and type 3 (WS3, OMIM # 148820). WS1 and WS3 are characterized by dystopia canthorum, a lateral displacement of inner canthus of the eyes. This feature is highly penetrant for WS1, can be objectively determined by measuring the W indexa [1]. Hearing loss, pigmentary disturbances of the iris, hair and skin are other common features of Waardenburg syndrome with variable penetrance. Clinically, Waardenburg syndrome can be diagnosed based on the presence of two major features or one major feature plus two minor features [2]. Major features are sensorineural hearing loss, iris pigmentary abnormality, hair hypopigmentation, dystopia canthorum (W index > 1.95) and affected first-degree relative. Minor features include skin hypopigmentation, synophrys, broad nasal root, hypoplastic alae nasi, and premature graying of the hair. Short stature is not associated with WS1. WS3, sometimes called Klein-Waardenburg syndrome, includes abnormalities of the upper limbs in addition to dystopia canthorum, hearing loss and changes in pigmentation. About 90% of pathogenic variants in PAX3 are nucleotide level alterations detectable by sequencing, whereas an additional 10% of WS1 are caused by large deletions involving part or the whole PAX3 gene [3]. These variants may be detected by karyotyping [4,5], FISH [6,7] and MLPA [8,9]. Recently chromosomal microarray (CMA) analysis has allowed for a more accurate delineation of the deletion interval and genes involved, allowing for better genotype-phenotype correlation study.
Case presentation
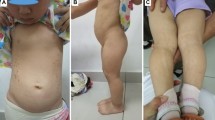
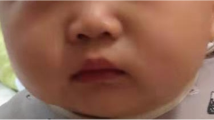
Here we report a 4-year and 6 month-old Chinese boy with a rare combination of Waardenburg syndrome type 1 and severe short stature. The patient was the first child of a non-consanguineous marriage. He had severe short stature (83.5 cm (−5.8SD)) and poor weight gain (10 kg (−4.1 SD)). Both parents had short stature but are healthy: father’s height is 153 cm (−3.2 SD) and mother’s height is 148 cm (−2.3SD). Patient’s short stature is proportionate. His facial features included dystopia canthorum (W index = 2.31), mild synophrys, slightly up-slanted palpebral fissure, posteriorly rotated ear, alae nasi hypoplasia and micrognathia (Figure 1a and b). He has normal intelligence and hearing. No heterochromia or other pigmentation anomalies was noticed. Physical examination did not find any other abnormalities. The patient met the clinical diagnostic criteria of WS1. Routine blood, urine tests as well as liver and renal function tests were all normal. Endocrine tests for TSH, FT3, FT4, FSH, LH and insulin were normal. Insulin-like growth factor-1 (IGF-1) was 52.7 ng/ml, lower than the normal range (55 ~ 452 ng/ml). The peak growth hormone level was 7.57 ng/ml at 90 minutes after taking moderate levodopa and pyridostigmine bromide. Brain MRI revealed a pituitary gland about 3.2 cm long without abnormal morphology. Bone age based on left hand X-ray is 1.5 years.

Characteristic facial feature of the patient. A and B are the front and side views of the patient. Notable features include ocular hypertelorism, dystopia canthorum, short philtrum and wide based nose root with hypoplasia alae nasi.
The patient underwent recombinant human growth hormone (rhGH) replacement therapy for over a year. The daily dosage was 0.11 IU/kg. Growth velocity and side-effect profile were monitored for at regular intervals (Table 1). The patient exhibited a good response to rhGH treatment. He showed a 9.2 cm/year growth rate and an improvement of 1 SD of height after one year treatment (Figure 2).

Height standard deviation curve during replacement therapy with rhGH.
CMA test of patient’s DNA using illumina Human SNP cyto-12 array revealed a 4.46 Mb de novo deletion at 2q35-q36.2 (chr2:221,234,146-225,697,363) (hg19, Figure 3). The deletion involved the whole PAX3 gene which is responsible for the Waardenburg syndrome phenotype and neighboring genes including EPHA4.

Array profile of the deleted region at 2q35q36.2. The left panel is the whole chromosome view; the right panel is a zoomed-in view of the deleted region demarcated by the two arrows and a vertical red bar.
We evaluated previously published cases of overlapping deletions with our case’s (Table 2 and Figure 4). We noticed that two thirds of deletion cases reported short stature or growth retardation as one of the phenotypic features when EPHA4 gene was involved in the deletions. The remaining cases did not provide height information. The animal model supported the notion that the EPHA4 deletion is responsible for short stature. The Spr mutant was created by X-ray mutagenesis and characterized by a cytogenetically detectable deletion of band C4 on mouse chromosome 1. Heterozygous mice displayed white spotting of the belly, tail and feet, equivalent pigmentary features of WS. They also had persistent growth retardation throughout their development [10]. The deletion is approximately syntenic to human chr2:218,449,525-232,459,056 region, both Pax3 and Epha4 were involved in the deletion. The other Splotch mutants caused by missense mutation (Spd) [11] or splicing mutation (Sp) [12] in Pax3 gene do not exhibit growth retardation, suggesting genes adjacent to Pax3 are responsible for the growth retardation phenotype. The most important evidence came from the recent epha4 knockout mouse model. Both heterozygous and homozygous epha4 knockout mice showed significant postnatal growth retardation in a dose dependent manner [13]. So far only one patient has been reported to carry a deletion involving only the EPHA4 gene (DECIPHER 282314). It was a de novo deletion. The available phenotype of this patient involves the shortening of digital bones such as short 1st metacarpal, short distal phalanx of hallux, short distal phalanx of the 2nd finger, short distal phalanx of the thumb, short distal phalanx of toe and short first metatarsal. All these phenotypes were absent from either of the parents. The height information was not available for this patient. Due to their close proximity on chromosome 2, EPHA4 and PAX3 are often co-deleted as the cases shown in Figure 4. Most of them had reported short stature as a clinical feature [4-6,8,9,14-17]. In addition to EPHA4 and PAX3, there are about 10 more OMIM genes involved at the deletion interval of our patient, three are associated with human diseases (AP1S3 is associated with the susceptibility to pustular psoriasis-15; MRPL44 is likely associated with Combined oxidative phosphorylation deficiency 16; CUL3 is associated with Pseudohypoaldosteronism, type IIE). None of these genes or the rest of the genes at the interval are known to be associated with short stature. EPHA4 is not currently a known disease causing gene. Our patient and other similar deletion patients, as well as the mouse model provide compelling evidence indicating EPHA4 as a novel short stature gene.

Previously published cases with overlapping deletion at 2q35-q36.2. (Each horizontal red bar represents the deletion interval of a case. The origins of these cases are listed in Table 2).
EPHA4 is a member of the EPH family of receptor tyrosine kinases. It was demonstrated that EPHA4 interacts directly with growth hormone receptor (GHR) and JAK2 [11]. They form a ternary complex in human cells [11]. It is conceivable that EPHA4 haploinsufficiency causes short stature by impairing the growth hormone pathway that regulates down-stream effectors such as STAT5B and IGF1. Segregation of the short stature phenotype with EPHA4 mutations will further support the causal relationship.
It is interesting to note that growth hormone injection did not improve the growth of the Epha4 homozygous knockout mouse. In our patient, growth hormone replacement therapy resulted in significant improvement of growth velocity and height. It is not known if heterozygous knockout mice will respond to growth hormone or not. The response we observed in our patient provided preliminary indication for rhGH treatment in patients with EPHA4 deletion.
Conclusions
The rare combination of Waardenburg syndrome phenotype and short stature observed in our patient can be explained by the haploinsufficiency of both PAX3 and EPHA4 genes involved within the deletion. Our analysis indicated that EPHA4 is a novel short stature gene. Recombinant human growth hormone treatment improved the height of the patient; suggesting that diagnosis may help in determining utility of growth hormone in other individuals with EPHA4 associated short stature.
Consent
We obtained a written consent from patient’s parent for utilizing patient‘s clinical information, genetic details and photos in this publication. The study is approved by the internal review board of the Guangxi Maternal and Child Health Hospital.
Endnotes
aW index is defined as X + Y + a/b. a = inner canthal distance; b = interpupillary distance; c = outer canthal distance. A W index result greater than 1.95 is abnormal.
Abbreviations
- PAX3:
-
Paired box 3
- WS:
-
Waardenburg syndrome
- CMA:
-
Chromosomal microarray
- EPHA4:
-
Ephrin Receptor A type 4
- SD:
-
Standard deviation
- TSH:
-
Thyroid stimulating hormone
- FT3:
-
Free triiodothyronine
- FT4:
-
Free thyroxine
- FSH:
-
Follicle stimulating hormone
- LH:
-
Luteinizing hormone
- IGF-1:
-
Insulin-like growth factor-1
- MRI:
-
Magnetic resonance imaging
- rhGH:
-
Recombinant human growth hormone
- EPH:
-
Ephrin
- GHR:
-
Growth hormone receptor
- JAK2:
-
Janus kinase 2
References
Arias S, Mota M. Apparent non-penetrance for dystopia in Waardenburg syndrome type I, with some hints on the diagnosis of dystopia canthorum. J Genet Hum. 1978;26(2):103–31.
Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher Jr JH, Beighton P, et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Am J Hum Genet. 1992;50(5):902–13.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391–406.
Tassabehji M, Newton VE, Leverton K, Turnbull K, Seemanova E, Kunze J, et al. PAX3 gene structure and mutations: close analogies between Waardenburg syndrome and the Splotch mouse. Hum Mol Genet. 1994;3(7):1069–74.
Pasteris NG, Trask BJ, Sheldon S, Gorski JL. Discordant phenotype of two overlapping deletions involving the PAX3 gene in chromosome 2q35. Hum Mol Genet. 1993;2(7):953–9.
Wu BL, Milunsky A, Wyandt H, Hoth C, Baldwin C, Skare J. In situ hybridization applied to Waardenburg syndrome. Cytogenet Cell Genet. 1993;63(1):29–32.
Lu-Kuo J, Ward DC, Spritz RA. Fluorescence in situ hybridization mapping of 25 markers on distal human chromosome 2q surrounding the human Waardenburg syndrome, type I (WS1) locus (PAX3 gene). Genomics. 1993;16(1):173–9.
Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MLPA in Waardenburg syndrome. Genet Test. 2007;11(2):179–82.
Matsunaga T, Mutai H, Namba K, Morita N, Masuda S. Genetic analysis of PAX3 for diagnosis of Waardenburg syndrome type I. Acta Otolaryngol. 2013;133(4):345–51.
Epstein DJ, Malo D, Vekemans M, Gros P. Molecular characterization of a deletion encompassing the splotch mutation on mouse chromosome 1. Genomics. 1991;10(1):89–93.
Vogan KJ, Epstein DJ, Trasler DG, Gros P. The splotch-delayed (Spd) mouse mutant carries a point mutation within the paired box of the Pax-3 gene. Genomics. 1993;7(2):364–9.
Epstein DJ, Vogan KJ, Trasler DG, Gros P. A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant. Proc Natl Acad Sci U S A. 1993;90(2):532–6.
Jing X, Miyajima M, Sawada T, Chen Q, Iida K, Furushima K, et al. Crosstalk of humoral and cell-cell contact-mediated signals in postnatal body growth. Cell Rep. 2012;2(3):652–65.
Warter S, Lausecker C, Pennerath A. A girl with a deletion (2) (q34q36): cytogenetic and clinical observations. Hum Genet. 1976;32(2):225–7.
Kirkpatrick SJ, Kent CM, Laxova R, Sekhon GS. Waardenburg syndrome type I in a child with deletion (2) (q35q36.2). Am J Med Genet. 1992;44(5):699–700.
Nye JS, Balkin N, Lucas H, Knepper PA, McLone DG, Charrow J. Myelomeningocele and Waardenburg syndrome (type 3) in patients with interstitial deletions of 2q35 and the PAX3 gene: possible digenic inheritance of a neural tube defect. Am J Med Genet. 1998;75(4):401–8.
Melnyk AR, Muraskas J. Interstitial deletion of chromosome 2 region in a malformed infant. Am J Med Genet. 1993;45(1):49–51.
Acknowledgements
We would like to thank the parents of our patient for their cooperation. The study is funded by the National Science and Technology Support Program of China for the standardization of Screening, Diagnosis and Treatment of inherited metabolic disorders (2012BAI09B04).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CL, XF, JL and JQ carried out the clinical evaluation and treatment of the patient. RC, JW and BX carried out the chromosomal microarray testing for the patient and his parents. YS and SC conceived of the study, and participated in its design and coordination. YS, SC and CL helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ information
YS is an ABMG-certified medical geneticist. SC, CL, XF, JQ and JL are board-certified clinical pediatricians. RC, JW and BX are molecular laboratory medical technologist.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Li, C., Chen, R., Fan, X. et al. EPHA4 haploinsufficiency is responsible for the short stature of a patient with 2q35-q36.2 deletion and Waardenburg syndrome. BMC Med Genet 16, 23 (2015). https://doi.org/10.1186/s12881-015-0165-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-015-0165-2