
Abstract
Background
The widespread use of antiretroviral therapy (ART) has resulted in the development of transmitted drug resistance (TDR), which reduces ART efficacy. We explored TDR prevalence and its associated risk factors in newly diagnosed individuals in Guangxi.
Methods
We enrolled 1324 participants who were newly diagnosed with HIV-1 and had not received ART at voluntary counselling and testing centres (VCT) in Guangxi, China, who had not received ART. Phylogenetic relationship, transmission cluster, and genotypic drug resistance analyses were performed using HIV-1 pol sequences. We analysed the association of demographic and virological factors with TDR.
Results
In total, 1151 sequences were sequenced successfully, of which 83 (7.21%) showed evidence of TDR. Multivariate logistic regression analysis revealed that there was significant difference between the prevalence of TDR and unmarried status (adjusted odds ratio (aOR) = 2.41, 95% CI: 1.23–4.71), and CRF08_BC subtype (aOR = 2.03, 95% CI: 1.13–3.64). Most cases of TDR were related to resistance to non-nucleoside reverse transcriptase inhibitors (4.87%) and V179E was the most common mutation detected. We identified a total of 119 HIV transmission clusters (n = 585, 50.8%), of which 18 (15.1%) clusters showed evidence of TDR (36, 41.86%). Three clusters were identified that included drug-resistant individuals having a transmission relationship with each other. The following parameters were associated with TDR transmission risk: Unmarried status, educational level of junior high school or below, and CRF08_BC subtype may be a risk of the transmission of TDR.
Conclusions
Our findings indicated that moderate TDR prevalence and highlighted the importance of continuous TDR monitoring and designing of strategies for TDR mitigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Guangxi is located in Southwest China, adjacent to Vietnam and the Yunnan and Guangdong provinces. It is one of the areas in China that is most heavily affected by the human immunodeficiency virus 1 (HIV-1) in China [1]. Since the identification of the first individual infected with HIV in Guangxi in 1996 [2], the number of infected individuals has increased. By the end of 2017, Guangxi has the second highest number of reported HIV cases (113,500) in the country [3]. The Free Antiretroviral Treatment Program was launched in 2003 in China, and the “Treat for All” policy was implemented in 2016, and under which HIV-positive individuals were treated after being diagnosis of HIV regardless of their CD4+ cell count [4].
The widespread use and increased coverage of ART has reduced significantly reduced the risk of HIV transmission and decreased HIV-related morbidity and mortality [5]. Meanwhile, the increase in ART access corresponds to an increase in HIV drug resistance, which can be transmitted to newly infected individuals. TDR in HIV has become a major concern as it may lead to the failure of first-line ART [6]. Certain studies have suggested that significant large variations in the prevalence of TDR can be expected in different areas worldwide based on the differences in the availability of treatments and variances in socioeconomic development [7,8,9]. The prevalence is relatively high in high-income countries; for e.g., it is 11.2% in the United States [10], 14.7% in Romania [11] and 9.9% in Spain [12]. In middle- and low-income countries, lower estimates of TDR the prevalence have been reported; for e.g., it is 6.3% in Latin America [8], 5.7% in India, and < 5.0% in African countries [13]. A nationwide cross-sectional survey conducted in 2015 revealed 3.6% overall prevalence of drug resistance to be in China [14]. However, more recently, the prevalence of TDR been reported to increase to 12.2% in Tianjin [15], 17.4% in Shanghai [16], and 6.12% in Beijing [17].
As the HIV epidemic continues to spread, it is essential to investigate the changing trends in HIV-1 genetics as well as the prevalence and transmission of TDR in individual locations. Here, we performed an extensive cross-sectional study on individuals newly diagnosed with HIV between 2016 and 2018 in Guangxi.
Methods
Study subjects and sample collection
Between January 2016 and December 2018, we enrolled 1324 individuals who were newly diagnosed (non-diagnosed earlier) with HIV-1 and had not received ART were enrolled at voluntary counselling and testing centres in Guangxi, China, who had not received ART. After obtaining the participants’ informed consent from the participants, we collected peripheral blood samples and epidemiological data. Plasma was separated within 12 h of blood collection and stored at − 80 °C until further use.
HIV-1 RNA extraction, amplification, and sequencing
Viral RNA was extracted from the plasma samples using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The target fragment of 1316 bp in the pol gene (HXB2: 2147–3462; encoding the protease and the first 299 residues of reverse transcriptase), which spans the reverse transcriptase and protease-encoding regions, was amplified using nested polymerase chain reaction (PCR) according to a previously described protocol [18]. PCR products of the correct size (1316 bp) were excised from the gel, purified using a gel extraction kit (Qiagen, Hilden, Germany), and sequenced on an ABI3730 sequencer (Applied Biosystems, Carlsbad, CA, USA) .
Phylogenetic analysis
We edited all sequences with Sequencher v5.1 software (Genecodes, Ann Arbor, MI) and aligned them using BioEdit 7.1 software (Ibis Biosciences, Carlsbad, CA, USA) [17]. In order to identify the subtype of the virus gene, all the subtyping reference sequences were downloaded from the Los Alamos HIV database. The reference sequences were selected based on the following criteria: 1) inclusion of the major HIV-1 subtypes and circulating recombinant forms (CRFs); 2) covered pol gene sequence; 3) primarily originating from China and countries adjacent to Guangxi. In the end, 117 reference sequences were included, which covering all subtypes in China. Neighbour-joining method, which was used for the identification of gene subtypes [4, 14, 16, 17], was used to generated phylogenetic tree based on the Kimura 2-parameter model with 1000 bootstrap replicates using the MEGA7.0 software (available at: http://www.megasoftware.net) [19].
Drug resistance analysis
We evaluated clinically relevant resistance to nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), or protease inhibitors (PIs) using the Stanford University HIV Drug Resistance Database Genotypic Resistance Interpretation Algorithm (version 8.8) and the International Antiviral Society Drug Resistance Mutation list [16]. The degree of drug resistance to each antiretroviral drug was categorised as susceptible, potential low-level resistance, low-level resistance, intermediate resistance, or high-level resistance.
Transmission cluster construction
The aligned sequences were analysed using the HyPhy software to calculate the genetic distance, and the Tamura-Nei 93 pairwise genetic distance was calculated for all pairs of sequences. A genetic distance of ≤1.5% between two sequences was considered to indicate potential transmission partners [14]. The data is converted into edge lists, and the network was constructed by identifying pairs of sequences (nodes) and their potential transmission relationships (edge) using the visualisation sofware Cytoscape 3.5.1. We described the characteristics of the network, including the number of sequences (nodes), links (edges), and clusters (groups of linked sequences) [20].
Statistical analysis
SPSS version 21.0 software (IBM, Chicago, IL, USA) was used for statistical analysis. The data in this study comprised categorical variables indicated with frequencies and percentages. Logistic regression analysis was performed to identify risk factors associated with TDR. A P-value < 0.05 was considered statistically significant. In case a P-value < 0.05 was obtained, the variable was included for further adjustment, otherwise, the variable was omitted from the adjustment.
Results
Distribution of HIV-1 subtypes and drug resistance
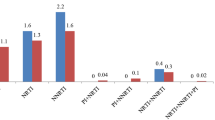
In our study subjects, CRF01_AE (42.14%) was the predominant subtype, followed by CRF07_BC (30.93%), CRF08_BC (15.90%), CRF55_01B (6.86%), and others (4.17%). The three main HIV-1 subtypes accounted for 88.97% of the cases among the newly diagnosed individuals. Other subtypes included the CRFs and unique recombinant forms CRF59_01B, CRF68_01B, CRF67_01B, and 85_BC, subtype A, B, and C. The overall prevalence of drug resistance was 7.21%, including 1.56% to PIs, 1.04% to NRTIs, and 4.87% to NNRTIs (Fig. 1).

Phylogenetic tree analysis based on the sequences of human immunodeficiency virus 1 pol gene. We constructed a phylogenetic tree based on the human immunodeficiency virus 1 pol gene sequences in the plasma samples of 1151 newly diagnosed individuals. CRF, circulating recombinant form; PI, protease inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, non-nucleoside reverse transcriptase inhibitor
Characteristics of the subjects and the factors associated with drug resistance
We enrolled 1324 HIV-1-infected individuals in the study, and we successfully sequenced and analysed the samples collected from 1151 (86.93%) individuals. The mean age was 40.8 years (range: 2–86 years). Most of the participants were single (46.39%), 41.62% were married, and 10.95% were divorced or widowed. Their level of educational was primarily junior high school or below (47.87%) or college (28.58%). Most of the individuals belonged to the ethnic majority Han (52.22%) and the Zhuang minority (41.96%). The major route of infection was sexual transmission; heterosexual transmission (60.21%), followed by MSM (35.19%), intravenous drug user (IDU, 4.0%) and mother-to-child transmission (MTCT, 0.6%). There was significant difference between the prevalence of TDR and marital status, subtype (all p values < 0.05) (Table 1).
Analysis of HIV Drug Resistance Mutation Sites (DRMs) with subtype
We identified 52 kinds of DRMs among the drug-resistant strains; 25 were associated with resistance to NNRTIs, 15 to NRTIs, and 12 to PIs. Individuals infected with the subtype CRF01_AE were the most likely to develop a PI-associated mutation, followed by those infected with CRF07_BC; the mutation sites primarily comprised M46V (0.35%) and Q58E (0.43%) mutations. Patients infected with the CRF01_AE subtype were the most likely to develop NRTI-associated mutations; the most common mutations were M41L (0.17%) and D67G (0.17%). Individuals infected with the subtypes CRF01_AE and CRF08_BC were the most likely to develop NNTRI-associated mutations; the most common mutations were V179E (7.21%), V179D (3.21%), and V106I (1.91%). The genotype CRF55_01B was the most likely to harbour V179E mutations (Fig. 2).

Analysis of drug resistance mutation sites with subtype
Level of resistance to different antiretroviral drugs
The PI-associated mutations were predicted to confer resistance to atazanavir (0.09%), darunavir (0.17%), fosamprenavir (0.26%), indinavir (0.09%), Kaletra (also known as lopinavir/ritonavir; 0.17%), nelfinavir (1.13%), saquinavir (0.14%), and tipranavir (0.61%). The NRTI-associated mutations were predicted to confer resistance to abacavir (0.52%), zidovudine (also known as azidothymidine; 0.52%), stavudine (0.78%), didanosine (0.43%), emtricitabine (0.35%), lamivudine (0.35%), and tenofovir (0.26%). The NNRTI-associated mutations were predicted to confer resistance to doravirine (2.87%), efavirenz (1.04%), etravirine (0.87%), nevirapine (1.22%), and rilpivirine (2.17%). The drug resistance level was mostly categorised as low (Table 2).
Drug resistance-associated transmission cluster analysis
We constructed an HIV-1 transmission network (Fig. 3). Of the 1151 subjects evaluated, 585 (50.8%) were segregated into 119 clusters, which included 490 men and 95 women. In the network, the cluster sizes ranged between 2 and 205; there were 96 (80.7%) clusters with size < 5, 14 (11.8%) clusters with 5 ≤ size < 10, and 9 (7.5%) clusters with size ≥10. The biggest cluster had 205 individuals (35.0%). (Fig. 3). We analysed the infection routes and found that 56.9% of the transmission cases occurred in heterosexual individuals, 40.7% in MSM, and 2.4% were of the IDU and MTCT. We also observed that 43.4% of TDR cases were included in the transmission network, and most (21, 58.3%) of which were primarily concentrated in four clusters. These TDR included 11 cases with resistance to PIs, 4 to NRTIs and 21 to NNRTIs, with the predominant gender being male (77.8%) and the major infection route being heterosexual transmission (83.3%). There were three clusters in which the TDR cases were connected with each other. We observed that there were four TDR females connected with 12 TDR males. To explore the factors associated with TDR, we performed bivariate and multivariate analyses. There was significant difference observed between the risk of TDR and unmarried status (aOR:4.24; 95%CI:1.51–11.9; P:0.006), level of junior high school or below (aOR:5.89; 95%CI:1.37–24.97; P:0.017), or infection with the CRF08_BC subtype (aOR:3.69; 95%CI:1.5–9.04; P:0.004) (Table 3).

Drug resistance within human immunodeficiency virus 1 transmission clusters. PI, protease inhibitor; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, non-nucleoside reverse transcriptase inhibitor; sensitive, sensitive to antiretroviral drugs
Discussion
We aimed to reveal the genetic characteristics and prevalence of transmitted HIV TDR among the newly diagnosed individuals in Guangxi, China. Our study revealed that the major epidemic HIV-1 subtypes detected in Guagnxi were CRF01_AE (42.14%), CRF07_BC (30.93%), CRF08_BC (15.9%) and CRF55_01B (6.86%). The distribution of HIV-1 subtypes has changed in the last two decades. Subtypes B and E were the major prevalent strains before 2000 [2]. In 2013, Liu et al. found that CRF01_AE (77.6%), CRF08_BC (10.7%), and CRF07_BC (7.4%) were the major strains in Guangxi [21]. In 2015, Zhang et al. observed that CRF01_AE (62.0%), CRF07_BC (25.0%), and CRF08_BC (6.5%) were the major strains [22]. However, the proportion of individuals infected with CRF01_AE gradually declined, whereas the proportion of individuals infected with CRF07_BC increased [21, 22]. CRF07_BC was first detected in intravenous-drug users in Guangxi in 2002 [23]. To our knowledge, in this study, we observed the first incidence of CRF68_01B, CRF85_BC, and unconfirmed unique recombinant strains in Guangxi. Their routes of transmission warrant further study. These findings indicated the existence of high genetic heterogeneity and subtype/CRF diversity in HIV-1 in Guangxi, and showed that the new CRFs had spread to provinces with population movement. Furthermore, the continuation of HIV recombination led to the production of new CRFs and URF, which made the HIV subtypes more diverse and complex. Additionally, the introduction of the new CRFs had a profound impact on the local HIV epidemic, and made the spread across a floating population easier. Therefore, the surveillance of HIV subtype should be strengthened further.
According to the categorisation method established by WHO [24], the overall prevalence of TDR in Guangxi was at a moderate level (5–15%). It was higher than the prevalence determined in the region in a previous study [25] as well as in other regions of China [17, 26, 27]. As the increase in TDR will affect the antiviral treatment and the spread of drug resistance, the surveillance of TDR should be strengthened, and measures to curb the increase of TDR should be adopted. A significant difference was observed between the prevalence of TDR and marital status and subtype, which contradicted from the results of a previous study [17, 28]. When marital status was used as a categorisation parameter, unmarried individuals were most likely to develop drug resistance. When categorised based on subtype, patients with the CRF08_BC subtype were the most likely to develop drug resistance. Therefore, these two high-risk factors should be considered in clinical settings. We found a significantly higher prevalence of mutations related to NNRTI resistance than of those associated with resistance to PIs or NRTIs. Mutations related to NNRTI resistance were common, especially V179E (7.21%) and V179D (3.21%). Most V179E mutations were detected in CRF55_01B, and most V179D mutations were detected in CRF01_AE, CRF07_BC, and CRF08_BC subtypes. In case of NNRTIs, the mutation V106I, which can cause low-level resistance to doravirine, was a major cause of drug resistance. High-level resistance to efavirenz and nevirapine primarily resulted from the mutations K103N, L100I, and P225H. In case of NRTIs, 0.86% of the individuals were predicted to be resistant to zidovudine, stavudine, and didanosine. In case of PIs, 1.74% of the individuals were predicted to be resistant to nelfinavir and tipranavir. Combinatorial therapy with lamivudine, efavirenz, and nevirapine has been prescribed as the first-line ART regimen in China, and the total drug resistance rate to these three drugs found to be 2.61% in our study. Therefore, the emergence of these mutations might be related to the use of this first-line regimen. The higher prevalence of TDR may be caused by prolonged ART, as noted in other studies [29, 30].
To further understand the transmission of drug resistance, we constructed transmission clusters based on HIV-1 sequences. Our cluster analysis revealed that 585 individuals could be segregated into 119 clusters, including MSM, heterosexual men, heterosexual women, and intravenous-drug users; this indicated that the transmission characteristics were complicated. In the largest cluster, which comprised 146 individuals (1 woman, 145 men), MSM was the major route of transmission. Analysis of this super cluster suggested that MSM were strongly associated with the local epidemic. Additionally, 43.4% (36/83) of the individuals infected by drug-resistant strains were included in 19 clusters, in which drug resistance was mostly associated with heterosexuality (88.89%). Three clusters were identified that included drug-resistant individuals sharing a transmission relationship with each other, which counted for 44.4% (16/36). The result indicated that the spread of TDR may have occurred in the transmission network. The major infection route in the three clusters was heterosexual transmission (83.3%), and there were five women carrying TDR included in the TDR transmission network. This shows that drug-resistant individuals, especially women should be studied further, because they may act as a potential source of TDR. Logistic regression analysis revealed that being unmarried, having educational level of junior high school or lower, and subtype CRF08_BC may be related to TDR to transmit in the clusters. The reasons for the association of these parameters with TDR to transmit should be investigated further.
Conclusions
The present study revealed a diverse, complex distribution of HIV-1 subtypes in Guangxi, and a moderate prevalence of TDR. However, there are several limitations in the study including the number of intravenous-drug users. Additionally, 13% of the samples could not be sequenced successfully, the major causes may include sample quality, low viral load in patients, or low sensitivity of reagents. The lack of data on contact between study participants was also another limitation. Future studies conducted on a larger, more diverse group with contact information available is necessary to design effective strategies for intervention in high-risk populations. Regardless, the risk of TDR was pronounced and must be factored into the treatment and prevention policies. Our findings will be helpful in determining the optimal primary ART and implementing effective interventions that target the major populations at risk in the area.
Availability of data and materials
The datasets are available from the corresponding author on reasonable request. Some of the sequences are accessible at GenBank (accession numbers, MW294209 - MW295352).
Abbreviations
- HIV:
-
Human immunodeficiency virus
- MSM:
-
men who have sex with men
- TDR:
-
Transmission drug resistance
- ART:
-
Antiretroviral therapy
- NNRTI:
-
Non-nucleoside reverse-transcriptase inhibitor
- NRTI:
-
Nucleoside reverse-transcriptase inhibitor
- PI:
-
Protease inhibitor
- EFV:
-
Efavirenz
- NVP:
-
Nevirapine
- FTC:
-
Emtricitabine
- 3TC:
-
Lamivudine
- ABC:
-
Abacavir
- DDI:
-
Didanosine
- D4T:
-
Stavudine
- TDF:
-
Tenofovir
- AZT:
-
Azidothymidine
- DRV/r:
-
Darunavir
- LPV/r:
-
Lopinavir
- ATV/r:
-
Atazanavir
- DRM:
-
Drug resistance mutation
- HIVDR:
-
HIV Drug Resistance Database
- TN93:
-
Tamura-Nei 93
- OR:
-
Odds ratio
- aOR:
-
Adjusted odds ratio
- CI:
-
Confidence interval
References
Ge XM, Yang WM, Zhu QY, Wu WL, Shen ZY, Zhu JH, et al. Epidemiological characteristics of HIV/AIDS in Guangxi Zhuang autonomous region, 2010-2017. Zhonghua Liu Xing Bing Xue Za Zhi. 2019;40:315–21.
Chen J, Young NL, Subbarao S, Warachit P, Saguanwongse S, Wongsheree S, et al. HIV type 1 subtypes in Guangxi Province, China, 1996. AIDS Res Hum Retrov. 1999;15:81–4.
Huang Y, Zhou O, Zheng Z, Xu Y, Shao Y, Qin C, et al. Effect of AIDS-defining events at initiation of antiretroviral therapy on long-term mortality of HIV/AIDS patients in Southwestern China: a retrospective cohort study. AIDS Res Ther. 2020;17:44.
Hao M, Wang J, Xin R, Li X, Hao Y, Chen J, et al. Low rates of transmitted drug resistances among treatment-naive HIV-1-infected students in Beijing. China AIDS Res Hum Retrovir. 2017;33:970–6.
Viviane DL, Richard H, David RB, Robert SH, Robert G, Benita YJ, et al. The combined effect of modern highly active antiretroviral therapy regimens and adherence on mortality over time. J Acquir Immune Defic Syndr. 2009;50:529–36.
Huang HY, Daar ES, Sax PE, Young B, Cook P, Benson P, et al. The prevalence of transmitted antiretroviral drug resistance in treatment-naïve patients and factors influencing first-line treatment regimen selection. HIV Med. 2008;9:285–93.
Pennings PS. HIV drug resistance: problems and perspectives. Infect Dis Rep. 2013;5:e5.
Frentz D, Boucher CAB, van de Vijver DA. Temporal changes in the epidemiology of transmission of drug-resistant HIV-1 across the world. AIDS Rev. 2012;14:17–27.
Yang WL, Kouyos RD, Böni J, Yerly S, Klimkait T, Aubert V, et al. Swiss HIV Cohort Study (SHCS). Persistence of transmitted HIV-1 drug resistance mutations associated with fitness costs and viral genetic backgrounds. PLoS Path. 2015;11:e1004722.
Readhead AC, Gordon DE, Wang Z, Anderson BJ, Brousseau KS, Kouznetsova MA, et al. Transmitted antiretroviral drug resistance in New York state, 2006-2008: results from a new surveillance system. PLoS One. 2012;7:e40533.
Temereanca A, Ene L, Mehta S, Manolescu L, Duiculescu D, Ruta S. Transmitted HIV drug resistance in treatment-naive Romanian patients. J Med Virol. 2013;85:1139–47.
Vega Y, Delgado E, Fernández-Garcia A, Cuevas MT, Thomson MM, Montero V, et al. Epidemiological surveillance of HIV-1 transmitted drug resistance in Spain in 2004-2012: relevance of transmission clusters in the propagation of resistance mutations. PLoS One. 2015;10:e125699.
Ssemwanga D, Lihana RW, Ugoji C, Abimiku A, Nkengasong J, Dakum P, et al. Update on HIV-1 acquired and transmitted drug resistance in Africa. AIDS Rev. 2015;17:3–20.
Zhao S, Feng Y, Hu J, Li Y, Zuo Z, Yan J, et al. Prevalence of transmitted HIV drug resistance in antiretroviral treatment naïve newly diagnosed individuals in China. Sci Rep. 2018;8:12273.
Zheng MN, Ning TL, Zhou N, Zhao X, Li L, Zhu JJ, et al. Transmitted drug resistance among HIV infected men who have sex with men in Tianjin, 2014-2017. Zhonghua Liu Xing Bing Xue Za Zhi. 2018;39:619–24.
Wang Z, Zhang M, Zhang R, Liu L, Shen Y, Wang J, et al. Diversity of HIV-1 genotypes and high prevalence of pretreatment drug resistance in newly diagnosed hiv-infected patients in Shanghai, China. BMC Infect Dis. 2019;19:313.
Song YX, Xin RL, Li ZC, Yu HW, Lun WH, Ye J, et al. Prevalence of transmitted drug resistance among HIV-1 treatment-naive patients in Beijing. Epidemiol Infect. 2018;146:339–44.
Li J, Feng Y, Shen Z, Li Y, Tang Z, Xiong R, et al. Author correction: HIV-1 transmissions among recently infected individuals in Southwest China are predominantly derived from circulating local strains. Sci Rep. 2018;8:17478.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Li X, Xue Y, Lin Y, Gai J, Zhang L, Cheng H, et al. Evolutionary dynamics and complicated genetic transmission network patterns of HIV-1 CRF01_AE among MSM in Shanghai, China. Sci Rep. 2016;6:34729.
Liu W, Liang SJ, Yang JY, Li JJ, Wang B, Chen LL, et al. Distribution of HIV-1 subtypes in Guangxi Zhuang autonomous region, 2008-2009. Zhonghua Liu Xing Bing Xue Za Zhi. 2013;34:53–6.
Zhang J, Shen ZY, Li Z, Liang SJ, He C, Liang FX, et al. Genetic characteristics of CRF01_AE among newly diagnosed HIV-1-infected 16- to 25-year olds in 3 geographic regions of Guangxi, China. Medicine. 2015;94:e894.
McClutchan FE, Carr JK, Murphy D, Piyasirisilp S, Gao F, Hahn B, et al. Precise mapping of recombination breakpoints suggests a common parent of two BC recombinant HIV type 1 strains circulating in China. AIDS Res Hum Retrov. 2002;18:1135–40.
Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One. 2009;4:e4724.
Li G, Liang S, Harrison TJ, Tang Z, Shen Z, Wang X, et al. Prevalence of transmitted HIV-1 drug resistance remains low in Guangxi, China, eight years after scale-up of highly-active antiretroviral therapy. Intervirology. 2014;57:270–6.
Han ZG, Zhang YL, Wu H, Gao K, Zhao YT, Gu YZ, et al. Prevalence of drug resistance in treatment-naive HIV infected men who have sex with men in Guangzhou, 2008-2015. Zhonghua Liu Xing Bing Xue Za Zhi. 2018;39:977–82.
Chen M, Jia MH, Ma YL, Luo HB, Chen HC, Yang CJ, et al. The changing HIV-1 genetic characteristics and transmitted drug resistance among recently infected population in Yunnan, China. Epidemiol Infect. 2018;146:775–81.
Wang X, Liu X, Li F, Zhou H, Li J, Wang Y, et al. Epidemiological surveillance of HIV-1 transmitted drug resistance among newly diagnosed individuals in Shijiazhuang, Northern China,2014–2015. PLoS One. 2018;13:e198005.
Tostevin A, White E, Dunn D, Croxford S, Delpech V, Williams I, et al. Recent trends and patterns in HIV-1 transmitted drug resistance in the United Kingdom. HIV Med. 2017;18:204–13.
Hattori J, Shiino T, Gatanaga H, Mori H, Minami R, Uchida K, et al. Characteristics of transmitted drug-resistant HIV-1 in recently infected treatment-naive patients in Japan. J Acquir Immune Defic Syndr. 2016;71:367–73.
Acknowledgements
We wish to thank all the staff members of the Center for Disease Control and Prevention who were involved in the study; this includes the staff at the Center for Disease Control and Prevention of Nanning. We are grateful to Liujiang Yin, Jiaxiao Jiang, Jianjun Li, Kang Li, and Shanfang Qin for their coordination of the project.
Funding
This study was supported by the Guangxi Bagui Honor Scholars, National Science and Technology Major Project (Grant number 2018ZX10732101–002), and the Guangxi Health and Family Planning Commission Self-Financing Research Program (Grant number Z20170134). The funding body had no role in the data collection, analysis, and interpretation of the verbal data, and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Data curation, X.W.P. and J.H.H. Formal analysis, X.W.P., X.J.Z., and X.L.W. Funding acquisition, X.W.P. and S.J.L. Investigation, X.W.P. and K.L.T. Methodology, X.W.P. and N.Y.F. Project administration, X.W.P., Z.Y.S., and S.J.L. Resources, K.L.T., Q.H.. Supervision, Z.Y.S. and S.J.L. Validation, X.W.P. Writing-original draft, X.W.P. Writing-review & editing, Q.Y.Z., and S.J.L.. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Review Committee of Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control. All methods in this study were performed in accordance with the approved guidelines. All participants provided written informed consent to participate in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pang, X., Tang, K., He, Q. et al. HIV drug resistance and HIV transmission risk factors among newly diagnosed individuals in Southwest China. BMC Infect Dis 21, 160 (2021). https://doi.org/10.1186/s12879-021-05854-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-021-05854-w