
Abstract
Background
Extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli are responsible for severe infections worldwide. Whereas their genotypic and pathogenic characteristics are not documented in Democratic Republic of Congo (DRC), recent studies conducted at the Bukavu General Hospital in the South Kivu province highlighted their high prevalence in extra-intestinal infections. Here we provide data on molecular characterization of ESBL producing-Escherichia coli isolates from patients with extra-intestinal infections at this provincial hospital.
Methods
Whole-genome sequencing was carried out on 21 of these ESBL-producing Extra-intestinal Pathogenic Escherichia coli (ExPEC) for analysis of phylogenomic evolution, virulence factor and antimicrobial resistance (AMR) genes. Data were compared to phylogenetically close genomes using Multi-Locus Sequence Typing and Single Nucleotide Polymorphism-based phylogenetic approaches.
Results
The distribution of E. coli sequence types (ST) was as follows: ST 131 (n = 7), ST405 (n = 4), ST410 (n = 2), and other STs (ST10, ST58, ST95, ST393, ST443, S617, ST648, and ST2450). All ST131 belonged to the O25b-ST131 pandemic clone. Unexpectedly, they harbored more virulence genes than their GenBank counterparts. IncF plasmid replicons included novel FIB 69, FII 105 and FII 107 alleles. ESBL-genes included the plasmid-mediated CTX-M-15 in all isolates, and the SHV-12 allele. Other AMR genes included blaOXA-1, blaTEM-1, as well as genes encoding resistance against aminoglycosides, quinolones, chloramphenicol, rifampicin, tetracyclines, sulfonamides and trimethoprim.
Conclusion
Current data confirm the clonal spread of ESBL-producing ST131 and ST405 clones in patients from South Kivu, and the acquisition of resistance and virulence genes. A closer survey of AMR and virulence should therefore be prompted in this high-risk area.
Similar content being viewed by others

Background
The worldwide increasing prevalence of infections caused by multidrug-resistant (MDR) Gram-negative bacteria constitutes a serious threat to global public health, due to their association with a high morbidity and mortality rate which is fueled by the limited availability of effective antibiotics [1,2,3]. ESBL production is by far the most important determinant of rapid AMR spread among Enterobacteriaceae [4,5,6]. The dissemination of ESBL-producing Enterobacteriaceae is due to clonal expansion [7] and/or plasmid transfer [8]. ESBLs encoding genes are often located on large plasmids which are transferred to other bacteria by conjugation, enabling them to become ESBL producers [9, 10]. Beside ESBL genes, plasmids often harbor genes of resistance to multiple classes of antibiotics that result in MDR [8]. Several studies from all continents have consistently shown that CTX-M-15-producing E. coli is one of the most prevalent ESBL-producing Enterobacteriaceae [11] and that the global dissemination of ESBL-producing E. coli is associated with specific clones harboring a plasmid carrying the ESBL CTM-X-15 gene, especially ST131 and ST405 [12]. The same observation was made in a few African countries where CTX-M-15-producing E. coli belonging to phylogenetic groups A and D were found in extra-intestinal infections [13,14,15,16,17,18]. However, data on genotypic characterization of ExPEC are still lacking in several sub-Saharan countries among which DRC, the second largest African country [6]. This lack of accurate assessment of virulent and MDR ExPEC isolates makes it impossible to unravel the mechanisms underpinning their spread, hence to raise awareness about the best practices in health professionals. In two recent studies carried out at a tertiary care hospital in the Eastern province of South Kivu (Fig. 1), we observed a high prevalence of ESBL-producing Enterobacteriaceae in urinary tract and bloodstream infections among which a majority of ESBL-producing E coli in collected isolates [19, 20]. In the current study, a set of 21 ESBL-producing E. coli isolates were analyzed by whole-genome sequencing (WGS). This data set was used both to assess the phylogenomic relationship of MDR ESBL-producing E. coli isolates from DRC with GenBank genomes of MDR ESBL-producing ExPEC collected in other regions of the world, and to characterize their virulence and antimicrobial resistance genetic markers.

Map of the Democratic Republic of Congo. The area where the study was performed is indicated by a black arrow
Methods
Bacterial strains
The set of 21 ESBL-producing E. coli isolates analyzed in the current study came from a collection of isolates assembled between 2012 and 2014 from inpatients at the Bukavu General Hospital, South Kivu (DRC, see Fig. 1). All isolates were phenotypically identified by standard biochemical tests including oxidase testing, triple sugar iron, imviC tests (Indole, Methyl Red, Voges-Proskaeur, and Citrate utilization), urease and motility.
Antimicrobial susceptibility testing
The susceptibility to 14 antimicrobial agents (i.e. amikacin, amoxicillin, amoxicillin-clavulanic acid, ampicillin, ceftazidime, ceftriaxone, cefepime, cefotaxime, cefuroxime, chloramphenicol, ciprofloxacin, imipenem, trimethoprim-sulfamethoxazole and tetracycline) was determined by the disk diffusion method according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines as updated in January 2017 [21]. Minimum Inhibitory Concentrations (MIC) for each of the 15 antimicrobial agents was determined after 16–20 h incubation on Mueller-Hinton plates inoculated with suspensions of isolates at a fixed density (0.5 to 0.6 McFarland standard), using E-test strips (BioMérieux, Marcy l’Etoile, France) according to the manufacturer’s recommendations. Additionally, isolates were tested for ESBL-production by the double-disk synergy method on Mueller-Hinton agar using ceftazidime and ceftriaxone placed at a distance of 20 mm apart from a disk containing amoxicillin plus clavulanic acid. A clear-cut enhancement of the inhibition in front of either ceftazidime and/or ceftriaxone disks towards the clavulanic acid-containing disk (also called “champagne-cork” or “keyhole”) was interpreted as positive for ESBL production [22]. E-test strips (BioMérieux, Marcy l’Etoile, France) were used for confirmation of ESBL production, following the manufacturer’s instructions. E. coli ATCC 35218 and Klebsiella pneumoniae ATCC 700603 strains were used as ESBL-negative and positive controls, respectively. In addition, isolates were tested for the presence of the beta-lactamase AmpC phenotype using cefoxitin-cloxacillin disk diffusion test as described previously [23].
Whole-genome sequencing
Whole-genome paired-end sequencing was performed using the MiSeq sequencer (Illumina, San Diego, CA, USA). Accordingly, genomic DNA (gDNA) from ESBL-producing E. coli was isolated using the EZ1 Advanced XL Biorobot and the tissue DNA kit (Qiagen, Hilden, Germany) with the Bacterial card, according to the manufacturer’s instructions. For each isolate, genomic DNA was quantified using Qubit® fluorometric quantitation (ThermoFisher Scientific, Oregon, USA) and normalized to 0.2 ng/μl. A standard Nextera XT library (Illumina, San Diego, USA) was constructed for each genome with 1 ng gDNA as recommended by the manufacturer. Briefly, gDNA was simultaneously fragmented and tagged with sequencing adapters in a single step using Nextera transposome (Nextera XT DNA Library Preparation Kit, Illumina, San Diego, USA). Tagmented DNA was then amplified (12-cycle PCR amplification) and cleaned up with AMPure beads. Nextera libraries were quantified using Qubit and the size profile was analyzed on 2100 Bioanalyzer using High sensitivity DNA assay kit (Agilent Technologies, Waldbronn, Germany). Fragments with size ranging from 828 to 1433 bases were generated. Libraries selected for sequencing were normalized to 1 nM and pooled. The 1 nM pooled library was denaturated and diluted prior to loading on a MiSeq paired-end 2 × 150 (MiSeq reagent kit V2 (300 cycles) or 2 × 300 base pairs (bp) (MiSeq reagent kit V3 (600 cycles) sequence run.
Bioinformatics analysis
Paired-end reads from each E. coli isolate were assembled de novo using the Spades v.3.11.1 algorithm [24] to generate a draft genome sequence for each isolate and quality assessment for genome assemblies was carried out using QUAST 4.5 [25]. Raw genome data have been submitted to the European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena) and are available under accession number ERS1812814-ERS1812829. MLST typing was performed on draft (n = 21) and on complete genomes of ExPEC from GenBank by using the E. coli MLST scheme developed by Achtman [26] and the home-made Pathogenomic R package (https://github.com/JeromeAmbroise/Pathogenomics). The latter was used to screen all draft (n = 21) and complete genomes of E. coli sharing the same STs with DRC isolates for the virulence factor genes described in ExPEC [27,28,29] and/or available in the Virulence Finder database (https://cge.cbs.dtu.dk/services/VirulenceFinder/) with a threshold of 95% identity and a minimum length of 80%. Concurrently, each draft genome was screened for the presence of AMR genes. The complete list of screened genes was drawn up from the MEGARes database (https://megares.meglab.org). In order to selectively identify AMR genes acquired through horizontal gene transfer, the list based on MEGARes data was restricted to genes that were also found in the ResFinder database (https://cge.cbs.dtu.dk/services/ResFinder/), using BLASTn. In addition, SNP-based AMR chromosomal determinants were identified using the ARIBA software [30] with the MEGARes database. Assembled contigs were further assessed for the presence of plasmid replicons using the plasmid multilocus sequence typing (pMLST) database [31]. The F plasmids were further categorized by the FAB (FII, FIA, FIB) formula using the replicon sequence typing (RST) scheme described by Villa [32]. The DNA sequences of novel FIB and FII replicons were submitted to the pMLST database curator (https://pubmlst.org/plasmid/) for the assignment of the ST. All drafts (n = 21) and ST-relevant complete genomes from GenBank were submitted to kSNP3.0 for SNP identification and Maximum Likelihood phylogenetic tree construction. This software performs SNP identification without genome alignment nor requirement for reference genomes. In parallel, WGS data were used to characterize E. coli isolates through the combination of four DNA gene markers (i.e. ArpA, chuA, yjaA and TSPE4-C2) as described by Clermont et al. [33]. In brief, E. coli draft genomes were screened for the presence of these four genetic markers, a combination thereof determining the phylogenetic clustered distribution of the isolates. Those isolates which belong to the B2 phylogenetic group were further screened for the ST131-O25b clone-specific silent SNPs in the E. coli pabB gene (C267T and G573A, accession number: CP015085) as previously described [34].
Results
Antimicrobial susceptibility patterns
All isolates (n = 21) were MDR ESBL-producing E. coli (Fig. 2). MIC data are provided in in Table 1. The majority of isolates displayed low susceptibility to amoxicillin, amoxicillin-clavulanic acid, ampicillin, ceftazidime, cefuroxime, ceftriaxone, cefotaxime, chloramphenicol, ciprofloxacin, imipenem, trimethoprim-sulfamethoxazole and tetracycline. In contrast, all and 19/21 DRC E. coli isolates were susceptible to imipenem and amikacin, respectively.

Heatmap of antimicrobial susceptibility profiles of DRC ESBL-producing ExPEC isolates. White, grey and black boxes: susceptible, intermediate and resistant to the corresponding antimicrobial drug. All DRC ESBL-producing ExPEC isolates retained susceptibility to imipenem, while displaying resistance to at least three of the following antimicrobial families: beta-lactams (including third and fourth generation cephalosporins), quinolones, sulfonamids, tetracycline and chloramphenicol. AKN: amikacin; AMX: amoxicillin; AMC: amoxicillin-clavulanic acid; AMP: ampicillin; CRO: ceftriaxone; CAZ: ceftazidime; CTX: cefotaxime; CEFUR: cefuroxime; CIP: ciprofloxacin; IMP: imipenem; SXT: trimethoprim-sulfamethoxazole; FEP: cefepime; CHL: chloramphenicol; TET: tetracycline, ESBL: Extended Spectrum Beta-Lactamase phenotype
Whole-genome sequencing and assembly
Computation of the total number of reads and quality metrics of the assemblies (Additional file 1) showed homogenous results with a good quality profile for all isolates.
Phylogenomic analysis
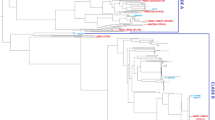
E. coli isolates (n = 21) were clustered into three major clades (Fig. 3). The first one grouped seven ST131 ExPEC isolates (DRC_BKV_03, DRC_BKV_04, DRC_BKV_05, DRC_BKV_12, DRC_BKV_13, DRC_BKV_14, and DRC_BKV_16), one ST95 isolate (DRC_BKV_19), and one ST648 isolate (DRC_BKV_20). All ST131 isolates belonged to the same sub-clade and to the phylogenetic group B2 according to Clermont et al. [33]. All displayed the C267T and the G573A substitutions in the pabB gene in line with their O25b-ST131 status [34] (Fig. 3). The second clade included seven isolates belonging to unrelated various STs (i.e., DRC_BKV_01: ST617; DRC_BKV_08: ST10; DRC_BKV_15: ST2450; DRC_BKV_17 and DRC_BKV_21: ST410; DRC_BKV_11: ST58; DRC_BKV_18: ST443).

Maximum Likelihood phylogenetic tree based on the SNP differences within the core genomes of DRC ESBL-producing ExPEC (in black) along with other well characterized counterparts from GenBank (in grey)
The third clade contained four ST405 isolates (i.e., DRC_BKV_06, DRC_BKV_07, DRC_BKV_09, and DRC_BKV_10), and one ST393 isolate (DRC_BKV_02).
Detection of virulence factors genes
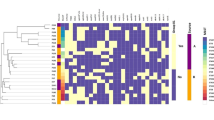
At first sight, virulence factors identified in DRC ST131 E.coli isolates are similar to those reported in pandemic CTX-M-15-producing E. coli O25b-ST131 [28, 35,36,37]. However, E. coli O25-b-ST131 from DRC harbored significantly (p < 0.01, t-test) more virulence genes (Fig. 4) as illustrated by the presence of the traT gene carried by all but one (DRC_BKV_12) DRC E.coli O25b-ST131. This gene was not detected in any of the E. coli genomes selected from Genbank (Fig. 4). Likewise, two out of 7 DRC O25b-ST131 isolates (i.e., DRC_BKV_04 and DRC_BKV_05) harbored the ireA virulence gene, which was absent from similar strain sequences in GenBank [35]. DRC ST131 E. coli were mostly (5/7) isolated from bloodstream and, as expected, harbored more virulence genes than DRC ST405 E. coli. The latter isolates were mostly (3/4) isolated from urine.

Virulence genes identified both in DRC ESBL-producing ExPEC (in dark grey for E. coli isolated from bloodstream, and in black for those isolated from urine samples), and in ExPEC from GenBank (in light grey). A gene was deemed present if it matched the reference sequence (minimal identity match of 95% with a minimal coverage of 80% of the gene sequence). * Virulence factor genes not extensively characterized in ExPEC as per previous reports [27,28,29], but described and/or available in the Virulence Finder database https://cge.cbs.dtu.dk/services/VirulenceFinder/)
Detection of AMR genes
Each draft genome sequence of ExPEC isolates from South Kivu harbored AMR genes. They consisted in chromosomal SNP-based determinants of AMR and/or plasmid-mediated AMR to various classes of antibiotics (Additional file 2 and Fig. 5). Some chromosomal SNP-based determinants of AMR corresponded to amino acid substitutions leading to resistance to several antibiotics, e.g. quinolones, sulfonamides, rifampicin, and elfamycins. Other chromosomal SNP-based determinants of AMR caused amino acids substitutions in several MDR genes (OMPF porin, PhoP multi-drug efflux pump) [38] and/or in genes which regulate the expression of several AMR genes, such as MARR (Multiple Antibiotic Resistance Regulator) and soxS (a member of Superoxide regulon) [39]. Analysis of MIC values for ciprofloxacin revealed that, whereas all DRC E. coli isolates were resistant to ciprofloxacin, high level resistance to this drug was overall associated with amino acid substitutions in quinolone-resistance-determining-regions (QRDR) of gyrA gene (S83 L, D87N) and/or in QRDR of parC gene (S80I, E84V, S57T, E84G). In addition, several SNPs resulting in amino acid substitutions were also characterized in gyrB and parE genes. Noticeably, none of these substitutions occurred in the respective QRDRs of both latter genes. These findings are consistent with other studies emphasizing the importance of substitutions in QRDRs of gyrA and parC proteins in the emergence of high level resistance to quinolones [40, 41]. However, given the limited set of data analyzed in this study, a confirmation of the role played by chromosomal SNP-based determinants in the emergence of quinolone resistance in DRC isolates requires further assessment.

Heatmap showing AMR genes found in the draft genome of DRC ESBL-producing ExPEC (n = 21) and present both in MEGARes and ResFinder. AMR genes are annotated as in MEGARes. The names of the strains indicated on the y axis are presented in the same order as in Figs. 2 and 3. 1. AMR genes for aminoglycosides: AAC3: Aminoglycoside N acetyltransferases AAC3; “ANT2”: Aminoglycoside O nucleotidyltransferases ANT2 DPRIME; “ANT3”: Aminoglycoside O nucleotidyltransferases ANT3 DPRIME; “APH3”: Aminoglycoside O phosphotransferases APH3-DPRIME; APH6: Aminoglycoside O phosphotransferases. 2. AMR genes for beta-lactams: CTX: Class A beta-lactamases CTX (in our study, all CTX genes consisted of CTX-M-15); TEM: Class A beta-lactamases TEM; CMY: Class C beta-lactamases CMY; OXA: Class D beta-lactamases OXA; PBP2: Penicillin binding protein PBP2. 3. AMR genes for quinolones: AAC(6′): Aminoglycoside N acetyltransferase (corresponds to AAC(6′)-Ib-cr-1in all DRC E. coli isolates); QNRB: Quinolone resistance protein Qnr QNRB. 4. AMR gene for macrolides: MPHA: Macrolide phosphotransferase MPHA. 5. AMR genes for phenicols: Chloramphenicol acetyltransferase CAT; Chloramphenicol acetyltransferases CATA; Chloramphenicol acetyltransferase CATB. 6. AMR genes for rifampicin: ARR: Rifampicin ADP ribosyltransferase Arr ARR. 7. AMR genes for sulfonamides: FOLP: Sulfonamide resistant dihydropteroate synthases FOLP; SULI: Sulfonamide resistant dihydropteroate synthases SULI; SULII: Sulfonamide resistant dihydropteroate synthases SULII. 8. AMR genes for tetracyclines: TETA: Tetracycline resistance major facilitator superfamily MFS efflux pumps TETA; TETB: Tetracycline resistance major facilitator superfamily MFS efflux pumps TETB; TETD: Tetracycline resistance major facilitator superfamily MFS efflux pumps TETD. 9. AMR gene trimethoprim: DHFR: Dihydrofolate reductase DHFR. It is of note that DRC_BKV_01 contained a “CTX-M” sequence as annotated in the MEGARes database. The BLAST analysis confirmed however that this annotation corresponded rather to the tnpA gene and not to CTX-M. DRC_BKV_01 is therefore reported as CTX-free in the figure
The CTX-M-15 gene, which encodes a protein responsible for the ESBL phenotype, was detected in all but one isolate (DRC_BKV_01). WGS-based analysis identified plasmid replicons in 21/21 isolates. Beside well-characterized alleles, RST revealed the presence of three new plasmid replicons, FIB 69 FII 105 and FII 107, which are reported here for the first time. Twelve different RST profiles were characterized in the 22 plasmid replicons: F105:A1:B69 (DRC_BKV_06, DRC_BKV_07, DRC_BKV_09, and DRC_BKV_10), F31:A4:B1 (DRC_BKV_01, DRC_BKV_08, and DRC_BKV_16), F48:A1:B49 (DRC_BKV_04 and DRC_BKV_05), F1:A2:B20 (DRC_BKV_13 and DRC_BKV_14), F2:A-:B1 (DRC_BKV_11 and DRC_BKV_15), F1:A1:B1 (DRC_BKV_18 and DRC_BKV_20), F1:A1:B16 (DRC_BKV_12), F36:A1:B1 (DRC_BKV_02), F1:A1:B49 (DRC_BKV_17), F107:A-:B:1 (DRC_BKV_19), F2:A1:B1 (DRC_BKV_21), and F1:A:2:B- (DRC_BKV_03). It is noteworthy that, except for IncF, no other incompatibility plasmid replicon types (i.e., IncA/C, IncH1, IncH2, IncI1, and IncN) were identified in these DRC isolates.
Discussion
WGS was used to analyze non-duplicated ESBL-producing E. coli isolates (n = 21) collected from patients at a tertiary care hospital in South-Kivu province of the DRC between 2014 and 2016. Despite the limited sample size, this study provides the first evidence that pandemic ESBL-producing E. coli O25b-ST131 and ST405 carrying blaCTX-M-15 are present in this DRC province, a factor that may be driving their widespread dissemination. Whereas establishing clonality between isolates sharing the same ST and high sequence similarity is difficult [37], data analysis of core genome, virulence and AMR genes supports the assumption that some of these isolates may have diverged recently from a common ancestor. The hypothesis of a common O25b-ST131 ancestor is supported by the perfect match between virulence and AMR genes as well as RST profiles of DRC_BKV_04 and DRC_BKV_05, with as few as 7 SNPs distinguishing their draft genome. It also applies to DRC_BKV_13 and DRC_BKV_14 whose core genomes only differed at 4 SNPs, as well as to the ST405 sub-clade (DRC_BKV_06, DRC_BKV_07, DRC_BKV_09 and DRC_BKV_10) which differed only by 1 SNP. These observations strengthen the hypothesis that local O25b-ST131 and ST405 sub-clades diverged recently from common ancestors.
Conversely, a markedly different virulence pattern rules out a clonal relationship between DRC_BKV_12 and Saudi E. coli isolate (accession n° CP015085) despite a close relatedness of their core genomes. Likewise, ST131 isolates from DRC do not seem to be closely related to other well characterized international isolates (i.e., NCTC13441: accession n° LT632320; uk_P46212: accession n° CP013658; EC958: accession n° HG941718) given their dissimilar virulence gene patterns.
Current data illustrate that ExPEC isolates from Bukavu, probably because of a permanent selective pressure of antibiotics, undergo a continuous remodeling process leading to spontaneous SNPs mutations and acquisition of virulence and AMR genes. This process may generate a genetic drift and/or shift from a common ancestor and the subsequent emergence of new clones. It is of note that E. coli belonging to other phylogenetic groups (A, B1) have also been isolated in extra-intestinal infections in other African countries [14, 15], but not yet in Western countries [27, 28].
To date, no convincing explanation for these discrepant observations can be put forward. It is however noteworthy that this study did not assess the clinical history of patients with infections caused by ESBL-producing bacteria, notably the effect of immune response due to HIV, malnutrition or other debilitating diseases. However, whether this may pave the way to severe ExPEC infections with non-B2 and non-D ESBL-producing E. coli requires confirmation.
Current WGS-based genotyping results corroborate our previous observations with ESBL-producing Enterobacteriaceae in urinary and bloodstream isolates in the South Kivu province [19, 20]. Extended set of virulence and AMR genes is expected to provide ESBL-producing E. coli strains capacity for surviving and thriving in their host and surrounding environment in presence of several antimicrobial agents [42]. The characterization of novel beta-lactamases and replicons suggest a high level of genetic plasticity within ExPEC plasmids carrying AMR genes. Moreover, the current irrational use of antibiotics in DRC is expected to facilitate nosocomial and community transmission and uncontrolled spread of these ESBL-producing ExPEC isolates.
Conclusions
Our results show that resistance of ESBL-producing E. coli to multiple classes of antibiotics in South Kivu Province of DRC is driven by several CTX-M-15 producing ST among which ST131 and ST405, as well as other STs considered not to be associated with ExPEC infections. These results corroborate previous observations on the staggering ability of pandemic clones O25b-ST131 and ST405 to adapt to new environmental conditions while also highlighting the continuous accumulation of both virulence and AMR genes in these pathogens. A vigorous approach through regional and international cooperation is needed to mitigate what looks like the inexorable spread of ESBL-producing E. coli in South Kivu province and beyond.
Abbreviations
- AMR:
-
Antimicrobial resistance
- DRC:
-
Democratic Republic of Congo
- ESBL:
-
Extended-spectrum beta-lactamase
- EUCAST:
-
European Committee on Antimicrobial Susceptibility Testing
- ExPEC:
-
Extra-intestinal Pathogenic Escherichia coli
- gDNA:
-
Genomic DNA
- MDR:
-
Multidrug-resistant
- MIC:
-
Minimum Inhibitory Concentrations
- MLST:
-
Multi-Locus Sequence Typing
- SNP:
-
Single Nucleotide Polymorphism
- ST:
-
Sequence Type
- WGS:
-
Whole-genome sequencing
References
Pitout JD, Laupland KB. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis. 2008;8(3):159–66.
Frieri M, Kumar K, Boutin A. Antibiotic resistance. J Infect Public Health. 2016;10:369–78.
Kaye KS, Pogue JM. Infections caused by resistant gram-negative Bacteria: epidemiology and management. Pharmacotherapy. 2015;35(10):949–62.
Canton R, Gonzalez-Alba JM, Galan JC. CTX-M enzymes: origin and diffusion. Front Microbiol. 2012;3:110.
Poirel L, Bonnin RA, Nordmann P. Genetic support and diversity of acquired extended-spectrum beta-lactamases in gram-negative rods. Infect Genet Evol. 2012;12(5):883–93.
Storberg V. ESBL-producing Enterobacteriaceae in Africa - a non-systematic literature review of research published 2008-2012. Infect Ecol Epidemiol. 2014;4:20342.
Nicolas-Chanoine MH, Bertrand X, Madec JY. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev. 2014;27(3):543–74.
Carattoli A. Plasmids and the spread of resistance. Int J Med Microbiol. 2013;303(6–7):298–304.
Rawat D, Nair D. Extended-spectrum beta-lactamases in gram negative Bacteria. J Glob Infect Dis. 2010;2(3):263–74.
Mshana SE, Hain T, Domann E, Lyamuya EF, Chakraborty T, Imirzalioglu C. Predominance of Klebsiella pneumoniae ST14 carrying CTX-M-15 causing neonatal sepsis in Tanzania. BMC Infect Dis. 2013;13:466.
Brolund A. Overview of ESBL-producing Enterobacteriaceae from a Nordic perspective. Infect Ecol Epidemiol. 2014;4:24555.
Naseer U, Sundsfjord A. The CTX-M conundrum: dissemination of plasmids and Escherichia coli clones. Microb Drug Resist. 2011;17(1):83–97.
Fam N, Leflon-Guibout V, Fouad S, Aboul-Fadl L, Marcon E, Desouky D, El-Defrawy I, Abou-Aitta A, Klena J, Nicolas-Chanoine MH. CTX-M-15-producing Escherichia coli clinical isolates in Cairo (Egypt), including isolates of clonal complex ST10 and clones ST131, ST73, and ST405 in both community and hospital settings. Microb Drug Resist. 2011;17(1):67–73.
Mshana SE, Imirzalioglu C, Hain T, Domann E, Lyamuya EF, Chakraborty T. Multiple ST clonal complexes, with a predominance of ST131, of Escherichia coli harbouring blaCTX-M-15 in a tertiary hospital in Tanzania. Clin Microbiol Infect. 2011;17(8):1279–82.
Aibinu I, Odugbemi T, Koenig W, Ghebremedhin B: Sequence type ST131 and ST10 complex (ST617) predominant among CTX-M-15-producing Escherichia coli isolates from Nigeria. In: Clin Microbiol Infect. vol. 18, 2011/12/24 edn; 2012: E49–E51.
Coque TM, Baquero F, Canton R. Increasing prevalence of ESBL-producing Enterobacteriaceae in Europe. Euro Surveill. 2008;13:19044.
Petty NK, Ben Zakour NL, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan MD, Gomes Moriel D, Peters KM, Davies M, et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci U S A. 2014;111(15):5694–9.
Rogers BA, Sidjabat HE, Paterson DL. Escherichia coli O25b-ST131: a pandemic, multiresistant, community-associated strain. J Antimicrob Chemother. 2011;66(1):1–14.
Irenge LM, Kabego L, Vandenberg O, Chirimwami RB, Gala JL. Antimicrobial resistance in urinary isolates from inpatients and outpatients at a tertiary care hospital in south-Kivu Province (Democratic Republic of Congo). BMC Res Notes. 2014;7:374.
Irenge LM, Kabego L, Kinunu FB, Itongwa M, Mitangala PN, Gala JL, Chirimwami RB. Antimicrobial resistance of bacteria isolated from patients with bloodstream infections at a tertiary care hospital in the Democratic Republic of the Congo. S Afr Med J. 2015;105(9):752–5.
EUCAST TECoAST-: Breakpoint tables for interpretation of MICs and zone diameters Version 7.1, valid from 2017-03-10. 2017, Version 7.0, valid from 2017-01-01:5.
Drieux L, Brossier F, Sougakoff W, Jarlier V. Phenotypic detection of extended-spectrum beta-lactamase production in Enterobacteriaceae: review and bench guide. Clin Microbiol Infect. 2008;14(Suppl 1):90–103.
Tan TY, Ng LS, He J, Koh TH, Hsu LY. Evaluation of screening methods to detect plasmid-mediated AmpC in Escherichia coli, Klebsiella pneumoniae, and Proteus mirabilis. Antimicrob Agents Chemother. 2009;53(1):146–9.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5.
Wirth T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006;60(5):1136–51.
Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. Coli infections in the United States. Clin Infect Dis. 2010;51(3):286–94.
Johnson TJ, Logue CM, Johnson JR, Kuskowski MA, Sherwood JS, Barnes HJ, DebRoy C, Wannemuehler YM, Obata-Yasuoka M, Spanjaard L, et al. Associations between multidrug resistance, plasmid content, and virulence potential among extraintestinal pathogenic and commensal Escherichia coli from humans and poultry. Foodborne Pathog Dis. 2012;9(1):37–46.
Koga VL, Tomazetto G, Cyoia PS, Neves MS, Vidotto MC, Nakazato G, Kobayashi RK. Molecular screening of virulence genes in extraintestinal pathogenic Escherichia coli isolated from human blood culture in Brazil. Biomed Res Int. 2014;2014:465054.
Hunt M, Mather AE, Sanchez-Buso L, Page AJ, Parkhill J, Keane JA, Harris SR. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genom. 2017;3(10):e000131.
Carattoli A, Zankari E, Garcia-Fernandez A, Voldby Larsen M, Lund O, Villa L, Moller Aarestrup F, Hasman H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–903.
Villa L, Garcia-Fernandez A, Fortini D, Carattoli A. Replicon sequence typing of IncF plasmids carrying virulence and resistance determinants. J Antimicrob Chemother. 2010;65(12):2518–29.
Clermont O, Christenson JK, Denamur E, Gordon DM. The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep. 2013;5(1):58–65.
Clermont O, Dhanji H, Upton M, Gibreel T, Fox A, Boyd D, Mulvey MR, Nordmann P, Ruppe E, Sarthou JL, et al. Rapid detection of the O25b-ST131 clone of Escherichia coli encompassing the CTX-M-15-producing strains. J Antimicrob Chemother. 2009;64(2):274–7.
Totsika M, Beatson SA, Sarkar S, Phan MD, Petty NK, Bachmann N, Szubert M, Sidjabat HE, Paterson DL, Upton M, et al. Insights into a multidrug resistant Escherichia coli pathogen of the globally disseminated ST131 lineage: genome analysis and virulence mechanisms. PLoS One. 2011;6(10):e26578.
Mshana SE, Falgenhauer L, Mirambo MM, Mushi MF, Moremi N, Julius R, Seni J, Imirzalioglu C, Matee M, Chakraborty T. Predictors of blaCTX-M-15 in varieties of Escherichia coli genotypes from humans in community settings in Mwanza, Tanzania. BMC Infect Dis. 2016;16:187.
Holmes A, Allison L, Ward M, Dallman TJ, Clark R, Fawkes A, Murphy L, Hanson M. Utility of whole-genome sequencing of Escherichia coli O157 for outbreak detection and epidemiological surveillance. J Clin Microbiol. 2015;53(11):3565–73.
Ziervogel BK, Roux B. The binding of antibiotics in OmpF porin. Structure. 2013;21(1):76–87.
Duval V, Lister IM. MarA, SoxS and Rob of Escherichia coli - global regulators of multidrug resistance, virulence and stress response. Int J Biotechnol Wellness Ind. 2013;2(3):101–24.
Hopkins KL, Davies RH, Threlfall EJ. Mechanisms of quinolone resistance in Escherichia coli and Salmonella: recent developments. Int J Antimicrob Agents. 2005;25(5):358–73.
Chung YS, Hu YS, Shin S, Lim SK, Yang SJ, Park YH, Park KT. Mechanisms of quinolone resistance in Escherichia coli isolated from companion animals, pet-owners, and non-pet-owners. J Vet Sci. 2017;18(4):449–56.
Overdevest I, Haverkate M, Veenemans J, Hendriks Y, Verhulst C, Mulders A, Couprie W, Bootsma M, Johnson J, Kluytmans J. Prolonged colonisation with Escherichia coli O25:ST131 versus other extended-spectrum beta-lactamase-producing E. coli in a long-term care facility with high endemic level of rectal colonisation, the Netherlands, 2013 to 2014. Euro Surveill. 2016;21:30376.
Acknowledgments
We gratefully acknowledge the technical assistance of Michèle Bouyer (Defense Laboratories Department) and Mrs. Olga Mineeva-Sangwo (CTMA). We also thank the technical staff of the Clinical Biology Department at the Bukavu General Hospital. The authors wish to express their gratitude to the EMBL-EBI team (Genome Campus, Hinxton, Cambridgeshire, CB10 1SD, UK) for providing ENA accession numbers, and to Mrs. Alexandra Carattoli, the pMLST database curator, for the assignment of the pMLST ST.
Funding
This project “Support for the improvement of detection and identification capacities of infectious agents in the province of South Kivu” (Appui à l’amélioration des capacités de détection et d’identification des agents infectieux Sud-Kivu) was funded by the Belgian Cooperation Agency through the 2012 PIC (Projet Inter-Universitaire de Coopération), grant n° PIC-MA-353 of the ARES (Académie de Recherche et d’Enseignement Supérieur).
It was also supported by a grant (MED-20) from the Department Management of Scientific & Technological Research of Defence (IRSD-RSTD; Royal High Institute for Defence).
Availability of data and materials
Raw genome data are available at the European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena) under accession number ERS1812814-ERS1812829.
Author information
Authors and Affiliations
Contributions
LMI, JA, RBC, BB, JFD, JLG have made substantial contribution to conception and design, acquisition, analysis and interpretation of data. LMI, JA, JLG have been involved in drafting the manuscript or revising it critically. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocols were approved by the Institutional Review Board (IRB) of the Université Catholique de Bukavu, and the Division Provinciale de la Santé (South-Kivu, DRC). A waiver of informed consent was granted by the IRB and the study complied with international guidelines on antibiotic surveillance studies. In order to ensure confidentiality, samples were analyzed anonymously. No ethical problem was encountered during this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Quality metrics of genome assemblies (QUAST 4.5). (XLS 36 kb)
Additional file 2:
Amino acid substitutions in chromosomal AMR genes, as identified using ARIBA software. Genes involved in quinolones AMR (gyrA, gyrB, parC and parE) are highlighted in grey. Amino acid substitutions in quinolone resistance-determining region (QRDR) of the above-cited genes are in bold. (XLS 41.5 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Irenge, L.M., Ambroise, J., Bearzatto, B. et al. Whole-genome sequences of multidrug-resistant Escherichia coli in South-Kivu Province, Democratic Republic of Congo: characterization of phylogenomic changes, virulence and resistance genes. BMC Infect Dis 19, 137 (2019). https://doi.org/10.1186/s12879-019-3763-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-019-3763-3