
Abstract
Background
Shortening the standard 6-month treatment for drug-susceptible pulmonary tuberculosis (DS-PTB) would be a major improvement for TB case management and disease control.
Methods
We are conducting a randomized, open-label, controlled, non-inferiority trial involving patients with smear-positive, newly diagnosed DS-PTB cases nationwide to assess the efficacy and safety of two 4.5- month regimens in comparison to the standard 6-month WHO recommended regimen. The regimen used in one experiment group is a 4.5-month fluoroquinolone-containing regimen, which consists of full course of levofloxacin, isoniazid (H), rifampin (R), parazinamid (Z) and ethambutol (E). Regimen used in the second experiment group includes 4.5-month full course of H, R, Z, E with levofloxacin removed. Patients in the control group, receive H, R, Z and E for 2 months, followed by 4 months of H and R. The primary endpoint is treatment failure or relapse within 24 month after treatment completion.
Discussion
Results from this trial along with other studies will contribute to the science of constructing a shorter, effective and safe regiment for TB patients.
Trial registration
The protocol has been registered on ClinicalTrials.gov on 2 September,2016 with identifier NCT02901288.
Similar content being viewed by others
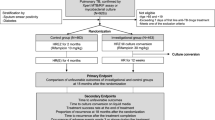

Background
Tuberculosis (TB) is now the leading infectious disease killer worldwide. In 2015, there were an estimated 10.4 million new TB cases worldwide. China is one of 30 TB high burden countries with third largest TB population globally. In 2015, 0.9 million new cases were estimated with an incidence of 67/100,000, accounting for 8.7% of the global total [1, 2]. Currently, 6-month short course remains the standard of care for the treatment of drug-susceptible pulmonary TB (DS-PTB), and its efficacy has been evaluated and established in controlled trials. Although standard 6-month regimens for drug- susceptible TB are highly efficacious, the duration of treatment is still considerably longer than that of most of respiratory infections. Poor adherence leads to treatment failure which in turn drives the emergence of drug resistance. Shortened treatment for DS-PTB would improve treatment outcomes and reduce costs. So regimens for TB that are shorter and/or simpler than the current 6-month regimen are needed.
The effectiveness of fluoroquinolones (FQ) in vitro, their sterilizing effect in both mice and human in vivo and their success in treating multi-drug resistant TB(MDR-TB) raised hope that if used as first line drugs [3,4,5,6], they might contribute to shortening therapy duration of DS-PTB. In 2014, 3 independent clinical trials named REMoxTB, RIFAQUIN and OFLOTUB/Gatifloxacin (Gfx) simultaneously reported final results of shortening treatment duration for DS-PTB by using FQ-based regimens [7,8,9]. Unfortunately, noninferiority for these regimens was not shown as compared to the standard 6-month regimen, indicating that shortening treatment to 4 months was not effective in these settings. Challenges as we faced, optimizing regimens for DS-PTB with shorter duration are still promising. The first pharmacodynamic data showed that levofloxacin(Lfx) was less effective than moxifloxacin(Mfx) and Gfx. Lfx was given at a dose less than 500 mg/day in these early studies [10]. With the prescription increase at the dose of 1000 mg/day in recent studies, Lfx showed the best early bactericidal activity as active as Mfx at 400 mg during the first 7 days of treatment [3]. Other studies have also shown that Mfx and Gfx are better than ofloxacin, but do not contain comparison with Lfx [11]. It seems to reveal that Lfx at high doses, rather than low doses, has better pharmacodynamic qualities than either Mfx or Gfx. Additionally, Lfx is less expensive, more widely available, and may be less likely to prolong the QT interval compared to later generation of FQs. Previous study showed that Z inhibited the ribosome-sparing process of trans-translation. Trans-translation is essential for freeing scarce ribosomes in nonreplicating organisms. Therefore, Z could eradicate persisting organisms which are the major cause of recurrence of TB [12]. If Z is given during the whole course, it can improve the sterilizing activities against mycobacterial tuberculosis (MTB) and reduce relapse of TB. On the basis of the supportive evidence, a multicenter randomized controlled clinical trial is currently in progress to evaluate whether 4.5 months of an Lfx-containing regimen will be as effective as 6 months of the standard regimen for the treatment of DS-PTB. Meanwhile, given the strong bactericidal and sterilizing activities of H, R, E and Z, parallel treatment shortening trial is also in progress with the consideration of increasing drug combination at continuation phase as compared with the standard 6-month regimen.
To our knowledge, preferable dosage and treatment duration of Lfx’s contribution combined with first line anti-TB drugs is not well studied. Neither is the shortening regimen with full length of treatment using H and R. This study aims to assess the efficacy and safety of 2 shortened regimens (study regimens) for DS-PTB compared to the WHO standardized 6-month regimen (control regimen). Success of the ultra-short course TB regimens would further shorten the treatment duration of DS-PTB, improve compliance and cure rates, reduce rates of adverse reactions and cost, as well as minimize the risk of drug resistance.
Methods and Design
Study design and oversight
We did a multicenter, randomized, controlled, open-label clinical trial within the framework of the China Tuberculosis Clinical Trial Consortium (CTCTC). The trial was sponsored by Ministry of Science and Technology of the People’s Republic of China and implemented by the leadership of Beijing Chest Hospital, Capital Medical University, together with other 34 TB specialized hospitals or Centers for TB Control and Prevention nationwide. The study protocol, the patient information sheets and consent forms were reviewed and approved by Central Ethics Committee of Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis and Thoracic Tumor Research Institute and the other participating medical facilities all approved the decision. The data and safety monitoring board planned to review efficacy and safety data at intervals of approximately 3 months throughout the study. Some of the trial medications were donated by Daiichi Sankyo Company Limited and Chengdu Jinhua Pharmaceutical Co.Ltd., but neither of these companies had any role in the study design, data accrual or further data analysis.
Study patients
Adult patients who are newly diagnosed, previously untreated or treated less than 1 month with anti-TB drugs, are determined by chest X-ray with any abnormality consistent with TB and positive results of sputum smears on two occasions, with culture-confirmed susceptibility to H, R, E and Z and Lfx. All patients with written or witnessed oral informed consent were recruited if they met the trial eligibility criteria at screening. Detailed inclusion and exclusion criteria is provided in Table 1.
Randomization and study regimens
Eligible patients who have provided written or witnessed oral informed consent were randomly assigned a unique study number sequentially on a web-based computerized algorithm, with the use of blocks of variable sizes which were stratified according to the study sites. Patients were randomized in a ratio of 1:1:1 to one of three regimens: a control regimen 2HREZ/4HR, which consists of 6 months of H and R administered daily, supplemented by E and Z in the first 2 months; a 4.5-month Lfx-containing regimen 4.5LfxHREZ, which is composed of H, E, Lfx, R, and Z administered daily, throughout the length of therapy; and another 4.5-month regimen4.5HREZ with Lfx removal from the former regimen. In all three groups, drug doses were adjusted according to patient’ weight, as described in Table 2. Treatment is directly observed at each local health facility and supervised by patient-friendly App management tools.
Sample size assumption
Under the conditions of the trial, we assumed that the standard 6-month regimen has a 95% or higher sputum culture conversion rate and with estimated 92.5% of that for 4.5-month study regimen based on the published data. The study was designed to determine whether either of the shortened study regimens was not inferior to the control regimen and a margin of noninferiority of 5% was used. With the assumption that as many as 20% of patients will loss to follow-up or not be assessable in the primary analysis, and with the consideration of approximately 5.5% primary drug resistance prevalence, a total target sample size was 3900 patients (1300 subjects per group) would be required to demonstrate noninferiority, with a statistical power of 0.8 and an alpha level of 0.05 (one-sided). Participant recruitment began in August 2016 and is expected to finish in December 2018.
Data management
All procedures conform to confidentiality standards for medical data. Authorized medical staff treating the patient is granted unlimited access to the patients’ data, whereas restricted access to anonymized data is granted to other staff and researchers. All data will be entered electronically into specific database developed by CTCTC for this study within 1 week and all original forms or records will be kept at all study sites. Participant trial folders are stored in order and in a secure and accessible place at each study site which will be maintained in storage for a period of 3 years after completion of the study. All data will be performed periodically on the CTCTC backup server.
Statistical analysis
According to the standard approach of noninferiority trials, the analysis will be conducted with both per-protocol (PP) and modified intention-to-treat (mITT) populations. Noninferiority, defined as a between-group difference of less than 5% in the upper boundary of one-sided 90% Wald confidence interval (which is equivalent to a one-sided significance of 5%) for the proportion of patients with primary unfavorable outcomes, must be shown on both analyses above to declare that either regimen is noninferior. We will use the chi-square test to compare the secondary outcomes of patients’ sputum-culture conversion at the end of 8 weeks (intensive phase) and treatment completion across treatment groups and the log-rank test to compare the time to culture-negative status. Other secondary outcomes will be analyzed with similar methods, including the time to an unfavorable outcome, the outcome at the end of treatment, adverse effect occurring frequency, patients’ adherence and the transition of radiological manifestation. Missing data will be dealt with the last observation carried forward imputation technique. Dropouts and withdrawals from the study will be recorded through the intervention and follow-up periods. When differences in baseline phenotype are present, these differences will be taken into account during analysis of treatment effect between groups, by regarding them as covariates.
Outcome measures
The primary efficacy outcome measures include the percentage of participants with treatment failure or relapse by 24 months after the end of treatment, percentage of participants with treatment failure at either 4.5 months or 6 months after randomization. The secondary efficacy and safety outcomes include the time to sputum (both smear and culture) conversion within intensive phase of 2 months and the percentage of participants with sputum conversion (both smear and culture) at the completion of treatment, the number of adverse reactions occurring on treatment and during the follow-up period, adherence to treatment and radiological manifestation transition. Of note, treatment failure was defined as sputum smear positive at the end of treatment and relapse after completion of treatment is defined as at least one positive culture in the scheduled follow-up visits in two years. Participants with negative cultures at the end of follow-up are considered to have had a favorable outcome. For all treatment failures and relapses, one culture of a sputum sample obtained before treatment and one culture of a sputum sample obtained after treatment failure or relapse are planned to store at −80 °C and the samples will be sent to national clinical TB reference lab in Beijing Chest Hospital for further research. The detailed observation schedule is present in Table 3.
Safety Reporting
Adverse events are defined and reported according to the Common Terminology Criteria for Adverse Events 4.03 (CTCAE). An adverse event is categorized as serious if it led to death, permanent or significant disability, a congenital anomaly or birth defect, life threatening, or required hospital admission for management. A data monitoring committee is needed with the consideration of the potential risk of this study at regular intervals during the trial. Interim analysis is planned when one third of enrolled patients have finished treatment. Thus efficacy and safety data of the study will be reviewed. The study can be stopped before reaching sample size if the data monitoring committee recommends termination of the study or termination of one of the treatment regimens due to unacceptable levels of treatment failure, relapse, adverse effect or mortality compared to the control arm. Important protocol modifications during this study will be updated to the trial registry online.
Clinical site monitoring and quality assurance
Family Health International 360 China office (FHI360) is responsible for monitoring data quality in accordance with trial Standard Operating Procedures (SOPs). Based on the monitoring plan, field visit and audit will be performed at different stages. All participant records, CRFs, and other source documents for the patients recruited in this study will be made available for review by the monitors. The investigator and staff are responsible for being present and available for consultation during scheduled site audit visits. A site field visit feedback will be sent to each study site made by authorized individuals. Investigators of each local site will be convened monthly via web-based remote conference system to share with the progress of study and discuss with the problems met during the trial searching for trouble shooting answers. Quality assurance (QA) procedures are carried out according to Quality Management documents. A review of these documents is undertaken by Quality Management Advisory Group (QMAG) of CTCTC. QMAG has the power to stop ongoing practices of any site which violates study protocol significantly and makes decision of its restarting or closing with respect to specific circumstance. Quality control (QC) procedures are performed at each study site by designated staff to guarantee all the practices comply with trial SOPs. Microbiology examinations are also under proper QC guideline developed by CTCTC. Moreover, Good Clinical Practice (GCP) training and appropriate Good Laboratory Practice (GLP) training will be provided for all staff involved in the trial; this will be a component of capacity strengthening of the trial.
Discussion
Current 6-month regimens for new smear positive DS-PTB are used in more than 90 countries and are designed and evaluated cost-effective [13]. With the exciting results of shortened regimens for MDR-TB cases from the previous 24 months to 9 months length of therapy [14–16], researchers have been paving the way to explore the probably shorter regimens for DS-PTB inferior to the current standard ones. In the 1970s, the introduction of R resulted in a 15 to 20% increase of sputum conversion at 8 weeks and shortening duration of treatment from 18 months to 9 months. The later introduction of Z led to a further 13% increase of sputum conversion at 8 weeks, allowing therapy to be further reduced from 9 months to 6 months [17–20]. Based on these findings, Medical Research Council (MRC) led the controlled clinical trial of five short-course (4-month) chemotherapy regimens for PTB patients including 2HRSZ/2HRZ, 2HRSZ/2HR, 2HRSZ/2HZ, 2HRSZ/2H, 2HRZ/2H. However, all the shortened regimens failed to present expected primary outcome, instead, with 16%, 11%, 32%, 30% and 40% recurrence after 24 months of treatment completion [21, 22]. For years with new drugs progress for TB chemotherapy, especially the favorable pharmacodynamics and bactericidal activity of FQ on MTB, shortening the treatment duration for DS-PTB by using a FQ-based regimen is of great hot topic 3 independent shortening clinical trials have been reported simultaneously with a failure to show noninferiority to the 6-month standard regimen attributed to the higher recurrence rate observed with the 4-month regimens, despite rapid culture conversion during treatment [7,8,9]. These findings raised questions that the shortening FQ-containing regimens did not work as adequately as the murine model did which may overpredict the sterilizing potency of the regimens.
Since Lfx administered with high dose is of comparable efficacy compared with Mfx and Gfx against MTB, it is possible to introduce Lfx to the regimen for shorter duration of DS-PTB. However, there is also concern that Lfx may fail as first-line anti-TB drugs because of its widely applied for common infections such as urinary and gastrointestinal tracts, wounds, and other lung infections. In high-TB-burden countries, a great number of cases with undiagnosed TB are likely to take an FQ, which could be selected for resistance in at least a fraction of MTB they harbor [23]. Several studies focus on FQ resistance and its association with previous FQ exposure indicated a low prevalence. Researchers from Korea found 2.6% FQ resistance in patients exposed to FQs while 3.4% resistance in patients without FQ exposure [24]. A study from Taiwan found no correlation of FQ resistance with either FQ exposure or duration of FQ exposure, but saw a positive correlation with previous anti-TB treatment and resistance to any other drugs [25]. Previous study from the same research team comparing sputum taken both before and after a course of FQs treatment showed that patients who received an FQ before standard anti-TB treatment had a poor prognosis, most likely as a result of the emergence of drug resistant. 11.1% of the MTB obtained FQ resistance after taking an FQ for 1–3 weeks [26]. The fifth national TB epidemiology survey in China of 2010 reported that Lfx resistant TB accounted for 9.1% among new TB patients [27]. A high prevalence of FQ-resistant TB may void the possibility of Lfx as first line drug for shortening DS-PTB treatment duration. Appropriate bacteriological and histopathological tests for TB should be performed as early as possible before Lfx-containing regimen is recommended. If the study regimen is successful, it is expected to provide a new standard of care for DS-PTB which would cut down the number of required clinic visits and the burden on the health care system and could also decrease the percentage of patients who fail to complete the full course of current longer therapy. If the study regimens are shown to be noninferior or superior to the control regimen, that would represent an even greater advance for patients with DS-PTB and TB control programmes globally. However, there are a few negative aspects that need to be considered before an FQ-containing first-line regimen could be broadly recommended. The FQs are fairly effective drugs for common nonspecific respiratory syndromes and community-acquired pneumonia, and curtailing this usage to ensure they remain effective as first-line TB drugs may not prove beneficial to all-cause morbidity and mortality at the community level. Also, using FQs as standard first-line therapy would reduce their effectiveness against MDR-TB, and could perhaps result in the emergence of more XDR-TB. Its implementation might be suggested only where the prevalence of Lfx-resistant TB is low and FQs are not routinely used for nonspecific respiratory symptoms when TB cannot be effectively excluded would reduce concerns above.
Abbreviations
- CTCTC:
-
China Tuberculosis Clinical Trial Consortium
- DS-PTB:
-
Drug-susceptible pulmonary tuberculosis
- E:
-
Ethambutol
- FHI360:
-
Family Health International 360
- FQ:
-
Fluoroquinolones
- GCP:
-
Good Clinical Practice
- Gfx:
-
Gatifloxacin
- GLP:
-
Good Laboratory Practice
- H:
-
Isoniazid
- Lfx:
-
Levofloxacin
- MDR-TB:
-
Multi-drug resistant TB
- Mfx:
-
Moxifloxacin
- MRC:
-
Medical Research Council
- MTB:
-
Mycobacterial tuberculosis
- QA:
-
Quality assurance
- QMAG:
-
Quality Management Advisory Group
- R:
-
Rifampin
- TB:
-
Tuberculosis
- Z:
-
Parazinamid
References
World Health Organization. Global tuberculosis report 2016. Geneva: WHO; 2016. WHO/HTM/TB/2016.13
Tiemersma EW, van der Werf MJ, Borgdorff MW, Williams BG, Nagelkerke NJ. Natural history of tuberculosis: duration and fatality of untreated pulmonary tuberculosis in HIV negative patients: a systematic review. PLoS One. 2011;6(4):e17601.
Peloquin CA, Hadad DJ, Molino LP, Palaci M, Boom WH, Dietze R, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52(3):852–7.
Drlica K, Malik M, Kerns RJ, Zhao X. Quinolone-mediated bacterial death. Antimicrob Agents Chemother. 2008;52(2):385–92.
Johnson JL, Hadad DJ, Boom WH, Daley CL, Peloguin CA, Elsenach KD, et al. Early and extended early bactericidal activity of levofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2006;10(6):605–12.
Rodriguez JC, Ruiz M, Lopez M, Royo G. In vitro activity of moxifloxacin, levofloxacin, gatifloxacin and linezolid against Mycobacterium tuberculosis. Int J Antimicrob Agents. 2002;20(6):464–7.
Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 2014;371(17):1577–87.
Merle CS, Fielding K, Sow OB, Gninafon M, Lo MB, Mthiyane T, et al. A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med. 2014;371(17):1588–98.
Jindani A, Harrison TS, Nunn AJ, Phillips PP, Churchyard GJ, Charalambous S, et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med. 2014;371(17):1599–608.
Takiff H, Guerrero E. Current prospects for the fluoroquinolones as first-line tuberculosis therapy. Antimicrob Agents Chemother. 2011;55(12):5421–9.
Rustomjee R, Lienhardt C, Kanyok T, Davies GR, Levin J, Mthiyane T, et al. A phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int J Tuberc Lung Dis. 2008;12(2):128–38.
Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE 3rd, et al. Pyrazinamade inhibits trans-translation in Mycobacterium tuberculosis. Science. 2011;333(6049):1630–2.
Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946–1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999;3(10):S231–79.
Nunn AJ, Rusen ID, Van Deun A, Torrea G, Phillips PP, Chiang CY, et al. Evaluation of a standardized treatment regimen of anti-tuberculosis drugs for patients with multi-drug-resistant tuberculosis (STREAM): study protocol for a randomized controlled trial. Trials. 2014;15:353.
Van Denu A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, et al. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med. 2010;182(5):684–92.
Aung KJ, Van Deun A, Declercq E, Sarker MR, Das PK, Hossain MA, et al. Successful “9-month Bangladesh regimen” for multidrug-resistant tuberculosis among over 500 consecutive patients. Int J Tuberc Lung Dis. 2014;18(10):1180–7.
East African/British Medical Research Council. Controlled clinical trial of short-course (6-month) regimens of chemotherapy for treatment of pulmonary tuberculosis. Lancet. 1972;1(7760):1079–85.
East African/British Medical Research Council. Controlled clinical trial of four short-course (6-month) regimens of chemotherapy for the treatment of pulmonary tuberculosis. Lancet. 1974;2(7889):1100–6.
East African/British Medical Research Council. Results at 5 years of a controlled comparison of a 6-month and a standard 18-month regimen of chemotherapy for pulmonary tuberculosis. Am Rev Respir Dis. 1977;116(1):3–8.
East African/British Medical Research Council. Controlled clinical trial of four short-course regimens of chemotherapy for two durations in the treatment of pulmonary tuberculosis. First report. Am Rev Respir Dis. 1978;118(1):39–48.
East African/British Medical Research Councils. Controlled clinical trial of five short-course (4-month) chemotherapy regimens in pulmonary tuberculosis: Second report of the 4th study. Am Rev Respir Dis. 1981;123(2):165–70.
East African/British Medical Research Council. Controlled clinical trial of five short-course (4-month) chemotherapy regimens in pulmonary tuberculosis. First report. Lancet. 1978;2(8085):334–8.
Ginsburg AS, Hooper N, Parrish N, Dooley KE, Doman SE, Booth J, et al. Fluoroquinolone resistance in patients with newly diagnosed tuberculosis. Clin Infect Dis. 2003;37(11):1448–52.
Park IN, Hong SB, Oh YM, Lim CM, Lee SD, Lew WJ, et al. Impact of short-term exposure to fluoroquinolones on ofloxacin resistance in HIV-negative patients with tuberculosis. Int J Tuberc Lung Dis. 2007;11(3):319–24.
Wang JY, Lee LN, Lai HC, Wang SK, Jan IS, Yu CJ, et al. Fluoroquinolone resistance in Mycobacterium tuberculosis isolates: associated genetic mutations and relationship to antimicrobial exposure. J Antimicrob Chemother. 2007;59(5):860–5.
Wang JY, Hsueh PR, Jan IS, Lee LN, Liaw YS, Yang PC, et al. Empirical treatment with a fluoroquinolone delays the treatment for tuberculosis and is associated with a poor prognosis in endemic areas. Thorax. 2006;61(10):903–8.
Wang Y, Xiao DL, Wang WJ, Liu X, Wang LX, Xu SF, et al. The fifth national epidemiological survey across China 2010. Beijing: Press of Military Medical Sciences; 2011. p. 169401. Chinese
Acknowledgements
The members of the Trial Management Group are as follows from 1Beijing Chest Hospital, Capital Medical University, Beijing, China and 2FHI360. Tang Shenjie1, Li Liang1, Gao Mengqiu1, Liu Yuhong1, Zhang Yao2, Du Jian1, Gao Jingtao1, Ma Liping1, Liu Rongmei1, Ma Yan1, Ding Xiaofen2,Zhong Li2, Xiao Hua, Shu Wei, Lv Zizheng, Guo Zhenyong, Wang Hong, Xie Shiheng.
The Trial Management Group would like to acknowledge the contribution of our site collaborators: Shi Junwei (The Sixth People’s Hospital of Nantong, Nantong, Jiangsu, China),Wang Quanhong (Taiyuan Fourth People’s Hospital, Taiyuan, Shanxi,China), Zhai Shuli (Sixth People’s Hospital of Nanyang, Nanyang, Henan, China), Shi Baosheng (The Infectious Disease Hospital of Hebi, Hebi, Henan, China), Peng Peng (Wuhan Institute For Tuberculosis Control, Wuhan, Hubei, China), Huang Yuping (Kaifeng Pulmonary Disease Hospital, Kaifeng, Henan, China), Sun Peng (Tuberculosis Hospital in Jilin Province, Changchun, Jilin, China), Fu Yanyong (Tianjin Centers for Disease Control and Prevention, Tianjin, China), Ji Binying (Harbin Chest Hospital, Harbin, Heilongjiang, China), Yan Xiaofeng (Chongqing Infectious Disease Medical Center, Chongqing, China),Pan Hongqiu (The Third People’s Hospital of Zhenjiang, Zhenjiang, Jiangsu, China), Wei Ming (Wuhan Medical Treatment Center,Wuhan, Hubei, China), Wu Guihui (Public Health Clinical Center of Chengdu, Chengdu, Sichuan, China), Liu Gang (Anhui Chest Hospital, Hefei, Anhui, China), Wang Jianyun (Pulmonary Hospital of Lanzhou, Lanzhou, Gansu, China), Fan Lin (Shanghai Pulmonary Hospital, Shanghai, China), Wu Meiying (The Fifth People’s Hospital of Suzhou, Suzhou, Jiangsu, China), Yan Xinglu (Heilongjiang Province Center for Tuberculosis Control and Prevention, Harbin, Heilongjiang, China), Liang Xuan (Shenyang Chest Hospital, Shenyang, Liaoning, China), Mei Zaoxian (Tianjin Haihe Hospital, Tianjin, China), Zhang Jian (Changchun Infectious Disease Hospital, Changchun, Jilin, China), A Ertai (Chest Hospital of Xinjiang Uygur Autonomous Region, Wulumuqi, Xinjiang, China), Liu Wen (Wangkai Infectious Disease Hospital of Zaozhuang, Tengzhou, Shandong, China), Tang Jintao (Infectious Disease Hospital in Heilongjiang Province, Harbin, Heilongjiang, China), Wu Huizhong (Centre for Tuberculosis Control of Guangdong Province, Guangzhou, Guangdong, China), Luo Ping (Beijing Research Institute for Tuberculosis Control, Beijing, China), Wu Qianhong (The Tuberculosis Prevention and Treatment Hospital of Shan Xi Province, Xi’an, Shanxi, China), Cui Xiuqin (The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, Henan, China), She Qilu(The 4th People’s Hospital of Qinghai Province, Xining, Qinghai, China), Chen Suliang (Hebei Province Center for Disease Prevention and Control, Shijiazhuang, Hebei, China), Huang Yisheng (Hunan Institute For Tuberculosis Control, Changsha, Hunan, China), Liu Feiying (Guangxi Center for Disease Prevention and Control, Nanning, Guangxi, China), Li Mingwu (The Third People’s Hospital of Kunming, Kunming, Yunnan, China), Ma Guoren (The Fourth People’s Hospital of Ningxia Autonomous Region, Yinchuan, Ningxia, China).
Availability of data and materials
Data of the study is collecting and will be deposited in publicly available repositories once completion.
Competing of interests
The authors declare that there are no actual or potential competing of interest in relation to this article.
Funding
Funding was provided by Key Project of Chinese National Programs (Grant No. 2015ZX10003001).
Authors’ contributions
TSJ is the Chief Investigator, and TSJ, LL and GMQ has conceived the initial trial concept and study protocol. TJS, GMQ, LYH and MLP have helped develop the trial design and protocol. DJ is the senior statistician and has written the statistical analytic plan and has carried out the power calculations. GJT, MFL and MLP are the trial managers who have contributed to the regulatory aspects of the trial. ZY provided monitoring and quality assurance of trial practice from each site. GJT drafted the original manuscript and all authors have read, contributed and approved the final manuscript.
Consent for publication
Because the protocol did not include any detail relating to individual patients, written informed consent for the publication is not applicable.
Ethics approval and consent to participate
The trial protocol, the patient information sheets and consent forms have been reviewed and approved by the Central Ethics Committee of Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis and Thoracic Tumor Research Institute (CEC Reference 2016–03-01).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gao, M., Gao, J., Du, J. et al. Efficacy of ultra-short course chemotherapy for new smear positive drug susceptible pulmonary tuberculosis: study protocol of a multicenter randomized controlled clinical trial. BMC Infect Dis 17, 435 (2017). https://doi.org/10.1186/s12879-017-2505-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-017-2505-7