
Abstract
Background
The study aimed to identify the potential biomarkers in pulmonary tuberculosis (TB) and TB latent infection based on bioinformatics analysis.
Methods
The microarray data of GSE57736 were downloaded from Gene Expression Omnibus database. A total of 7 pulmonary TB and 8 latent infection samples were used to identify the differentially expressed genes (DEGs). The protein-protein interaction (PPI) network was constructed by Cytoscape software. Then network-based neighborhood scoring analysis was performed to identify the important genes. Furthermore, the functional enrichment analysis, correlation analysis and logistic regression analysis for the identified important genes were performed.
Results
A total of 1084 DEGs were identified, including 565 down- and 519 up-regulated genes. The PPI network was constructed with 446 nodes and 768 edges. Down-regulated genes RIC8 guanine nucleotide exchange factor A (RIC8A), basic leucine zipper transcription factor, ATF-like (BATF) and microtubule associated monooxygenase, calponin LIM domain containing 1 (MICAL1) and up-regulated genes ATPase, Na+/K+ transporting, alpha 4 polypeptide (ATP1A4), histone cluster 1, H3c (HIST1H3C), histone cluster 2, H3d (HIST2H3D), histone cluster 1, H3e (HIST1H3E) and tyrosine kinase 2 (TYK2) were selected as important genes in network-based neighborhood scoring analysis. The functional enrichment analysis results showed that these important DEGs were mainly enriched in regulation of osteoblast differentiation and nucleoside triphosphate biosynthetic process. The gene pairs RIC8A-ATP1A4, HIST1H3C-HIST2H3D, HIST1H3E-BATF and MICAL1-TYK2 were identified with high positive correlations. Besides, these genes were selected as significant feature genes in logistic regression analysis.
Conclusions
The genes such as RIC8A, ATP1A4, HIST1H3C, HIST2H3D, HIST1H3E, BATF, MICAL1 and TYK2 may be potential biomarkers in pulmonary TB or TB latent infection.
Similar content being viewed by others


Background
Pulmonary tuberculosis (TB) is a widespread and fatal infectious disease. It is caused by various strains of mycobacteria, usually Mycobacterium tuberculosis [1]. It is estimated that one third of the world’s population are infected with M. tuberculosis [2]. More than 90 % of infected individuals remain asymptomatic with a latent infection [3]. With aging or immune system deteriorating, M. tuberculosis can reactivate and cause severe pulmonary TB [4]. Roughly 10 % of the latent infections can progress to active TB. The general signs and symptoms of this disease include fever, chills, night sweats, loss of appetite, weight loss, and fatigue [5]. Approximately, there are 9 million newly diagnosed cases of pulmonary TB and 1.5 million deaths annually, mostly in developing countries [6]. Therefore, uncovering therapeutic biomarkers in pulmonary TB would supply new insights for the diagnosis and treatment of this disease.
Numerous studies have been done to investigate the potential biomarkers for the treatment of pulmonary TB. For example, the serum CA-125 level is found significantly higher in active pulmonary TB than in inactive TB or normal sample, suggesting that CA-125 may be a beneficial parameter in determination of pulmonary TB activity [7]. Pollock et al. [8] suggested that M. tuberculosis Rv1681 protein was a diagnostic marker of active pulmonary TB. Additionally, Chowdhury et al. [9] reported that the serum interleukin (IL)-6 level of the active pulmonary TB patients following anti-tuberculosis drug therapy played an important role in immune-protection of the host against M. tuberculosis infection. Although many factors have been found, the diagnostic efficiency of pulmonary TB is still unsatisfactory [10]. Therefore, it is necessary to identify novel potential therapeutic biomarkers in pulmonary TB.
In the present study, the microarrays data GSE57736 were downloaded to identify the differentially expressed genes (DEGs) between pulmonary TB and latent tuberculosis infection samples. This dataset is deposited by Guerra-Laso et al. [11], the study of whom demonstrates that IL-26 is a candidate gene for TB susceptibility. In this study, we aimed to use different bioinformatics method to identify the DEGs between the two kinds of samples. Based on the obtained DEGs, we performed protein-protein interaction (PPI) network construction and network-based neighborhood scoring analysis. Besides, the hierarchical clustering analysis, functional enrichment analysis, correlation analysis and logistic regression analysis of DEGs were performed as well. Findings of this study may help to explore potential targets for the diagnosis and treatment in pulmonary TB.
Methods
Affymetrix microarray data
The array data of GSE57736 based on the platform of GPL13497 (Agilent-026652 Whole Human Genome Microarray 4x44K v2) was downloaded from Gene Expression Omnibus database, which was deposited by Guerra-Laso et al. [11]. The dataset available in this analysis contained 15 peripheral blood samples from seven pulmonary TB patients and eight latent tuberculosis infections. Among the seven pulmonary TB patients, there were three men and four women (average 82.7 years) with different clinical conditions: psoriasis (one patient), previous heart failure (one patient), arterial hypertension (two patients), bronchial asthma (one patient), chronic obstructive pulmonary disease (two patients), and prostate cancer (one patient). The eight latent tuberculosis infection samples included six men and two women (average 81.1 years), which had scored a positive result in the QuantiFERON-TB Gold in-tube test (Cellestis, Carnegie, Vic., Australia).
Data preprocessing and differential expression analysis
The probe IDs were converted into corresponding gene symbols based on the annotation information on the platform. When multiple probes corresponded to a same gene, the average expression value was calculated to represent the gene expression level. The limma package [12] in R was used to identify DEGs between pulmonary TB and TB latent infection samples. The Benjamin and Hochberg (BH) [13] method was used to adjust the raw p-values [false discovery rate (FDR)]. Then, log2-fold change (log2FC) was calculated. Only genes with |log2FC| > 1.0 and FDR < 0.05 were selected as DEGs.
PPI network construction
Human Protein Reference Database (HPRD, http://www.hprd.org/) [14] is a database of curated proteomic information pertaining to human proteins. Search Tool for the Retrieval of Interacting Genes (STRING, http://string.embl.de/) [15] is an online database which collects comprehensive information of proteins. In our study, the DEGs were mapped into STRING and HPRD databases to identify significant protein pairs with confidence score > 0.4. Then the PPI network was constructed based on these protein pairs using Cytoscape software [16].
Network-based neighborhood scoring
Neighborhood scoring [17] is a local method for prioritizing candidates based on the distribution of DEGs in the network. Gene in PPI network was assigned a score, which was based on its FC and the FC of its neighbors. The score of each node in PPI network was calculated with the neighborhood scoring method [18]. When the hub node and its neighborhood nodes were significantly highly expressed, the score > 0; When the hub node and its neighborhood nodes were significantly lowly expressed, the score < 0. Therefore, the top 50 nodes with higher scores and the last 50 nodes with lower scores were identified as important genes.
In order to confirm the efficiency of these important genes differentiating pulmonary TB and TB latent infection samples, hierarchical clustering analysis of the important genes was performed using cluster software [19]. The results were presented by TreeView software [20]. The expression profile data were filtered and normalized using cluster software. In detail, genes that were expressed in at least 80 % of the samples were selected. Besides, the genes and samples were normalized with median center method [21].
Functional enrichment analysis
Gene Ontology (GO, http://www.geneontology.org) database [22] is a collection of a large number of gene annotation terms. Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.ad.jp/kegg/) knowledge database [23] is applied to identify the functional and metabolic pathway. Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/) [24] is a tool that provides a comprehensive set of functional annotation for large list of genes. In this study, the important genes were performed GO and KEGG pathway enrichment analyses with DAVID. With the enrichment threshold of p-value < 0.05, the DEGs enrichment results in GO terms and KEGG pathways were obtained.
Correlation analysis
The immune system dysfunction has been suggested to play an important role in the occurrence of pulmonary tuberculosis [25]. Therefore, it is necessary to analyze the correlations of genes associated with immune system. In the present study, the Pearson correlation coefficient (PCC) was calculated across 15 samples to investigate potential regulatory relationships between important genes. The gene pairs with |PCC| > 0.5 were selected for further analysis.
Logistic regression analysis
In order to identify the risk biomarkers of pulmonary TB, we performed multivariate logistic regression analysis for the gene pairs with significant correlations (|PCC| > 0.5) using SPSS 19.0 software (SPSS Inc., Chicago, Illinois, USA) [26]. The genes with p-value < 0.05 were selected as feature genes.
Results
Identification of DEGs
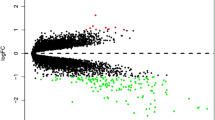
Based on the thresholds of |log2FC| > 1.0 and FDR < 0.05, a total of 1084 DEGs were identified between pulmonary TB and TB latent infection samples, including 565 down-regulated genes and 519 up-regulated genes. The result was shown in volcano plot (Fig. 1).

Volcano plot for the differentially expressed genes (DEGs). The x-axis represents the log2-fold change (log2FC). The y-axis represents the -log10 p-value. Blue-colored nodes are DEGs with p-value < 0.05 and |log2FC| > 1. Green-colored nodes are non-DEGs
PPI network construction
The PPI network consisted of 768 interaction pairs among 446 genes, including 253 down- and 193 up-regulated genes (Fig. 2). In this network, the proteins proto-oncogene tyrosine-protein kinase Fyn (FYN, degree = 34), CREB binding protein (CREBBP, degree = 28), growth factor receptor-bound protein 2 (GRB2, degree = 23) and guanine nucleotide binding protein (G protein) and beta polypeptide 2-like 1 (GNB2L1, degree = 21) were selected as hub nodes (genes) for the high connectivity degree.

The protein-protein interaction (PPI) network of differentially expressed genes (DEGs). The green nodes stand for down-regulated genes. The red nodes stand for up-regulated genes
Network-based neighborhood scoring
The top 50 nodes with higher scores and the last 50 nodes with lower scores were selected and the top 5 and last 5 genes were shown in Table 1. For instance, sirtuin 5 (SIRT5), tyrosyl-tRNA synthetase (YARS), sphingomyelin phosphodiesterase 2, and neutral membrane (SMPD2) had scores > 0. While the scores of solute carrier family 6 (neutral amino acid transporter), member 17 (SLC6A17), SCL6A8 and chloride channel, voltage-sensitive 7 (CLCN7) were < 0. Additionally, the down-regulated genes RIC8 guanine nucleotide exchange factor A (RIC8A), basic leucine zipper transcription factor, ATF-like (BATF) and microtubule associated monooxygenase, calponin LIM domain containing 1 (MICAL1), and the up-regulated genes ATPase, Na+/K+ transporting, alpha 4 polypeptide (ATP1A4), histone cluster 1, H3c (HIST1H3C), histone cluster 2, H3d (HIST2H3D), histone cluster 1, H3e (HIST1H3E) and tyrosine kinase 2 (TYK2) were also important genes.
Furthermore, hierarchical clustering analysis for these important genes showed that these 100 important genes could differentiate the pulmonary TB samples and TB latent infection samples (Fig. 3).

Clustering analysis of the important genes. The above dendrogram shows clustering of the samples. The red color stands for up-regulated genes, while green color stands for down-regulated genes
Functional enrichment analysis
GO enrichment analysis was carried out for the important genes. The significant (p < 0.05) GO biological process (BP) terms of up- and down-regulated genes were shown in Table 2 (p-values in ascending order). The down-regulated genes were significantly enriched in regulation of osteoblast differentiation (p = 0.00816) and positive regulation of hydrolase activity (p = 0.018904). Besides, the up-regulated genes were mainly related to nucleoside triphosphate biosynthetic process (p = 0.003512), mRNA export from nucleus (p = 0.004389) and purine nucleoside triphosphate metabolic process (p = 0.005796).
In addition, 2 pathways were enriched by the up-regulated important genes (Table 2), including adipocytokine signaling pathway and cardiac muscle contraction pathway. However, the down-regulated genes were not enriched in any pathways.
Correlation analysis
A total of 950 gene pairs were identified in Pearson correlation analysis. The top 10 highly correlated gene pairs were shown in Table 3. Specially, the expression levels of the top 4 correlated gene pairs (PCC > 0.9) were shown in Fig. 4, that was, RIC8A-ATP1A4, HIST1H3C-HIST2H3D, HIST1H3E-BATF and MICAL1-TYK2, besides, all of them showed positive correlations.

The expression levels of top 4 gene pairs. The x-coordinate represents samples; y-coordinate represents gene expression values. The blue lines represent RIC8 guanine nucleotide exchange factor A (RIC8A), histone cluster 2, H3d (HIST2H3D), tyrosine kinase 2 (TYK2) and histone cluster 1, H3e (HIST1H3E), respectively. The green lines represent ATPase, Na+/K+ transporting, alpha 4 polypeptide (ATP1A4), histone cluster 1, H3c (HIST1H3C), microtubule associated monooxygenase, calponin LIM domain containing 1 (MICAL1) and basic leucine zipper transcription factor, ATF-like (BATF), respectively. R stands for Pearson correlation coefficient
Logistic regression analysis
In order to identify the risk biomarkers of pulmonary TB, the gene pairs with significant correlations (|PCC| > 0.5) were performed logistic regression analysis. The analysis identified 80 significant feature genes, such as ATP1A4 (p = 0.031), RIC8A (p = 0.035), HIST1H3E (p = 0.005), BATF (p = 0.021), TYK2 (p = 0.008) and MICAL1 (p = 0.011). The prediction accuracy for the two groups of samples were 100 %.
Discussion
In this study, a total of 1084 DEGs including 565 down- and 519 up-regulated genes were selected. The up-regulated genes were mainly related to nucleoside triphosphate biosynthetic process. The down-regulated genes were significantly enriched in regulation of osteoblast differentiation. The gene pairs RIC8A-ATP1A4, HIST1H3C-HIST2H3D, HIST1H3E-BATF and MICAL1-TYK2 were identified with highly positive correlations. Besides, they were selected as feature genes in logistic regression analysis.
RIC8A encoding protein interacts with guanine nucleotide binding protein (G protein) [27]. It has been reported that RIC8A controls Drosophila neural progenitor asymmetric division by regulating heterotrimeric G proteins [28]. G protein is an important signal transducing molecule in cells [29], which activates MAP kinase signaling [30]. Elkington et al. [31] have reported that active pulmonary TB can be mediated by MAP kinase signaling pathway. In this study, RIC8A was down-regulated in pulmonary TB, suggesting that RIC8A may be associated with the pulmonary TB development through regulating MAP kinase signaling pathway with G proteins.
ATP1A4 is a member of P-type cation transport ATPase family and belongs to Na, K-ATPase subfamily. The P-type ATPases remove Ca2+ against very large concentration gradients in eukaryotic cells and play an important role in intracellular calcium homeostasis [32]. Importantly, calcium homeostasis involves in apoptosis and regulates important cellular events triggered upon infection of macrophages with pathogenic mycobacteria [33]. It has been reported that the M. tuberculosis blocks the delivery of the Na, K-ATPases [34]. Additionally, Rao et al. [35] have shown that de novo ATP synthesis is essential for the viability of hypoxic nonreplicating mycobacteria, requiring the cytoplasmic membrane to be fully energized. Interestingly, ATP1A4 was found enriched in nucleoside triphosphate biosynthetic process in this study. Therefore, we speculated that ATP1A4 may play a vital role in the occurrence of pulmonary TB by controlling ATP synthesis.
HIST1H3C, HIST2H3D and HIST1H3E belong to histone H3 family, which are responsible for controlling the dynamics of the chromosomal fiber in eukaryotes by regulating histone acetylation. Importantly, this process is essential in modulating gene transcription through chromatin organization, and perturbation of this process can result in aberrant gene transcription and cause some diseases, including lung diseases [36, 37]. Additionally, histones play a central role in DNA repair and DNA replication [38]. Boshoff et al. [39] reported that DNA-damaging agents were rich in vivo produced by host cells due to an effort to eradicate the M. tuberculosis. Therefore, DNA repair-related histones may an play important role in inhibiting M. tuberculosis infection. In this study, the up-regulation of HIST1H3C, HIST2H3D and HIST1H3E may be associated with occurrence of pulmonary TB.
BATF belongs to the adaptor-related protein 1 (AP-1)/activating transcription factor (ATF) superfamily of transcription factors. AP-1 family transcription factors control the differentiation of lymphocyte cells in immune system [40]. Lymphocytes are crucial in the immune defence against M. tuberculosis, which can secrete interferons (ITFs) in response to M. tuberculosis infection [41]. It has been reported that ITF-γ, a product of T lymphocytes, contributes to protective immunity against M. tuberculosis by activating macrophages in pulmonary TB [42]. Taken together, although the role of BATF in pulmonary TB has not been studied, we speculate that BATF may be involved in the occurrence of TB via immune system.
TYK2 encodes a tyrosine kinase belonging to Janus kinases (JAKs) family. It has been reported that JAKs are activated following interactions between cytokines and their cognate receptors on cell surface [43]. TYK2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting ITF-γ-dependent IL-10 reactivation [43]. Redford et al. [44] have reported that IL-10 can suppress the functions of macrophage and dendritic cell, which were required for the capture, control and initiation of immune responses to M. tuberculosis. Therefore, TYK2 may involve in the pathogenesis of pulmonary TB via regulating IL-10. For MICAL1, it can act as a cytoskeletal regulator [45]. Specially, cell migration and phagocytosis are critically dependent on cytoskeletal rearrangements [46]. It has been reported that cell migration and phagocytosis are important for resistance against pulmonary TB [47]. Therefore, the down-regulation of MICAL1 may be related to pulmonary TB via controlling cell migration and phagocytosis.
Conclusions
In conclusion, the present study identified several key gene pairs (RIC8A-ATP1A4, HIST1H3C-HIST2H3D, HIST1H3E-BATF, MICAL1-TYK2) associated with pulmonary TB or TB latent infection by comprehensive bioinformatics methods, which may provide new insights for the diagnosis and treatment of this disease. However, this study had some limitations. On the one hand, the sample size was small which might cause a high rate of false positive results. Secondly, there was no experimental verification. Therefore, further genetic and experimental studies with larger sample sizes are still needed to confirm the findings in this study.
Abbreviations
- AP-1:
-
Adaptor-related protein 1
- ATF:
-
Activating transcription factor
- ATP1A4 :
-
ATPase, Na+/K+ transporting, alpha 4 polypeptide
- BATF :
-
Basic leucine zipper transcription factor, ATF-like
- BH:
-
Benjamin and Hochberg
- BP:
-
Biological process
- CLCN7 :
-
Chloride channel, voltage-sensitive 7
- CREBBP :
-
CREB binding protein
- DAVID:
-
Database for annotation, visualization and integrated discovery
- DEGs:
-
Differentially expressed genes
- FYN :
-
Tyrosine-protein kinase Fyn
- G protein:
-
Guanine nucleotide binding protein
- GNB2L1 :
-
Guanine nucleotide binding protein and beta polypeptide 2-like 1
- GO:
-
Gene Ontology
- GRB2 :
-
Growth factor receptor-bound protein 2
- HIST1H3C :
-
Histone cluster 1, H3c
- HIST1H3E :
-
Histone cluster 1, H3e
- HIST2H3D :
-
Histone cluster 2, H3d
- HPRD:
-
Human protein reference database
- ITFs:
-
Interferons
- JAKs:
-
Janus kinases
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MICAL1 :
-
Microtubule associated monooxygenase, calponin LIM domain containing 1
- PCC:
-
Pearson correlation coefficient
- PPI:
-
Protein-protein interaction
- RIC8A :
-
RIC8 guanine nucleotide exchange factor A
- SIRT5 :
-
Sirtuin 5
- SLC6A17:
-
Solute carrier family 6neutral amino acid transporter), member 17
- SMPD2 :
-
Sphingomyelin phosphodiesterase 2, and neutral membrane
- STRING:
-
search tool for the retrieval of interacting genes
- TB:
-
Pulmonary tuberculosis
- TYK2 :
-
Tyrosine kinase 2
- YARS :
-
Tyrosyl-tRNA synthetase
References
Thakur M. Global Tuberculosis Control: WHO report 2010. National Medical J India. 2012;14(3):189–90.
van Pinxteren LAH, Cassidy JP, Smedegaard BHC, Agger EM, Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. Euro J Immunol. 2000;30(12):3689-98.
Vynnycky E, Fine PE. Lifetime risks, incubation period, and serial interval of tuberculosis. Am J Epidemiol. 2000;152(3):247–63.
Wu CY, Hu HY, Pu CY, Huang N, Shen HC, Li CP, Chou YJ. Pulmonary tuberculosis increases the risk of lung cancer. Cancer. 2011;117(3):618–24.
Fan S, Zhou G, Shang P, Meng L. Clinical Study of 660 Cases of Pulmonary Tuberculosis. Harbin Medical Journal. 2014;34(1):1-11.
Akachi Y, Zumla A, Atun R. Investing in improved performance of national tuberculosis programs reduces the tuberculosis burden: analysis of 22 high-burden countries, 2002–2009. Journal of Infectious Diseases. 2012;2(10):284-92. jis189.
Şahin F, Yildiz P. Serum CA-125: biomarker of pulmonary tuberculosis activity and evaluation of response to treatment. Clinical Investigative Med. 2012;35(4):E223–E8.
Pollock NR, Macovei L, Kanunfre K, Dhiman R, Restrepo BI, Zarate I, Pino PA, Mora-Guzman F, Fujiwara RT, Michel G. Validation of Mycobacterium tuberculosis Rv1681 protein as a diagnostic marker of active pulmonary tuberculosis. J Clin Microbiol. 2013;51(5):1367–73.
Chowdhury IH, Ahmed AM, Choudhuri S, Sen A, Hazra A, Pal NK, Bhattacharya B, Bahar B. Alteration of serum inflammatory cytokines in active pulmonary tuberculosis following anti-tuberculosis drug therapy. Mol Immunol. 2014;62(1):159–68.
Kanunfre KA, Leite OHM, Lopes MI, Litvoc M, Ferreira AW. Enhancement of diagnostic efficiency by a gamma interferon release assay for pulmonary tuberculosis. Clin Vaccine Immunol. 2008;15(6):1028–30.
Guerra‐Laso JM, Raposo‐García S, García‐García S, Diez‐Tascón C, Rivero‐Lezcano OM. Microarray analysis of Mycobacterium tuberculosis‐infected monocytes reveals IL‐26 as a new candidate gene for tuberculosis susceptibility. Immunology. 2014;144(2):291–301.
Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3(1):1-28.
Benjamini YH Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc Series B (Methodological). 1995;57:289–300.
Prasad TK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A. Human protein reference database—2009 update. Nucleic Acids Res. 2009;37 suppl 1:D767–D72.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39 suppl 1:D561–D8.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi:10.1101/gr.1239303.
Nitsch D, Gonçalves JP, Ojeda F, De Moor B, Moreau Y. Candidate gene prioritization by network analysis of differential expression using machine learning approaches. BMC Bioinform. 2010;11(1):460.
Emig D, Ivliev A, Pustovalova O, Lancashire L, Bureeva S, Nikolsky Y, Bessarabova M. Drug target prediction and repositioning using an integrated network-based approach. PLoS One. 2013;8(4):e60618.
Petushkova NA, Pyatnitskiy MA, Rudenko VA, Larina OV, Trifonova OP, Kisrieva JS, Samenkova NF, Kuznetsova GP, Karuzina II, Lisitsa AV. Applying of hierarchical clustering to analysis of protein patterns in the human cancer-associated liver. PLoS One. 2014;9(8):e103950. doi:10.1371/journal.pone.0103950.
Zhai Y, Tchieu J, Saier Jr MH. A web-based Tree View (TV) program for the visualization of phylogenetic trees. J Mol Microbiol Biotechnol. 2002;4(1):69–70.
Di Pietro C, Di Pietro V, Emmanuele G, Ferro A, Maugeri T, Modica E, Pigola G, Pulvirenti A, Purrello M, Ragusa M, Scalia M, Shasha D, Travali S, Zimmitti V. AntiClustal: Multiple Sequence Alignment by antipole clustering and linear approximate 1-median computation. Proc IEEE Comput Soc Bioinform Conf. 2003;2:326–36.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics. 2005;6(1):60.
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35 suppl 2:W169–W75.
Sotnichenko SA, Markelova EV, Skliar LF, Gel’Tser BI. Immune mechanisms of comorbidity of HIV infection and pulmonary tuberculosis. Ter Arkh. 2009;81(11):17–21.
Shek DT, Ma C. Longitudinal data analyses using linear mixed models in SPSS: concepts, procedures and illustrations. Scientific World Journal. 2011;11(1):42-76.
Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. J Biol Chem. 2003;278(10):8356–62. doi:10.1074/jbc.M211862200.
Wang H, Ng KH, Qian H, Siderovski DP, Chia W, Yu F. Ric-8 controls Drosophila neural progenitor asymmetric division by regulating heterotrimeric G proteins. Nat Cell Biol. 2005;7(11):1091–8.
Servin JA, Campbell AJ, Borkovich KA. G protein signaling components in filamentous fungal genomes. Biocommunication of Fungi. Springer; 2012. p. 21–38.
F-j S, Ramineni S, Hepler JR. RGS14 is a multifunctional scaffold that integrates G protein and Ras/Raf MAPkinase signalling pathways. Cell Signal. 2010;22(3):366–76.
Elkington PT, Emerson JE, Lopez-Pascua LD, O’Kane CM, Horncastle DE, Boyle JJ, Friedland JS. Mycobacterium tuberculosis up-regulates matrix metalloproteinase-1 secretion from human airway epithelial cells via a p38 MAPK switch. J Immunol. 2005;175(8):5333–40.
Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H + −ATPase to inhibit phagosome acidification. Proc Natl Acad Sci. 2011;108(48):19371–6.
McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. Biochem Biophys Res Commun. 1997;239(2):357–66.
Poirier V, Av-Gay Y. Mycobacterium tuberculosis modulators of the macrophage’s cellular events. Microbes Infect. 2012;14(13):1211–9.
Rao SPS, Sylvie A, Lucinda R, Thomas D, Kevin P. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci. 2008;105(105):11945–50.
Barnes P, Adcock I, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25(3):552–63.
Huang C, Sloan EA, Boerkoel CF. Chromatin remodeling and human disease. Curr Opin Genet Dev. 2003;13(3):246–52.
Peterson CL, Laniel M-A. Histones and histone modifications. Curr Biol. 2004;14(14):R546–R51.
Boshoff HI, Reed MB, Barry III CE, Mizrahi V. DnaE2 Polymerase Contributes to In Vivo Survival and the Emergence of Drug Resistance in Mycobacterium tuberculosis. Cell. 2003;113(2):183–93.
Schraml BU, Hildner K, Ise W, Lee W-L, Smith WA-E, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S. The AP-1 transcription factor Batf controls TH17 differentiation. Nature. 2009;460(7253):405–9.
El-Ahmady O, Mansour M, Zoeir H, Mansour O. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Annals Clin Biochem. 1997;34(2):160–4.
Sharma S, Bose M. Role of cytokines in immune response to pulmonary tuberculosis. Asian Pac J Allergy Immunol. 2001;19(3):213–9.
Shaw MH, Freeman GJ, Scott MF, Fox BA, Bzik DJ, Belkaid Y, Yap GS. Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-γ-dependent IL-10 reactivation. J Immunol. 2006;176(12):7263–71.
Redford P, Murray P, O’Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011;4(3):261–70.
Vanoni MA, Vitali T, Zucchini D. MICAL, the flavoenzyme participating in cytoskeleton dynamics. Int J Mol Sci. 2013;14(4):6920–59.
Fenteany G, Glogauer M. Cytoskeletal remodeling in leukocyte function. Curr Opin Hematol. 2004;11(1):15–24.
Leemans JC, Florquin S, Heikens M, Pals ST, van der Neut R, van der Poll T. CD44 is a macrophage binding site for Mycobacterium tuberculosis that mediates macrophage recruitment and protective immunity against tuberculosis. J Clin Investigation. 2003;111(5):681–9.
Acknowledgements
None.
Funding
None.
Availability of data and materials
The array data of GSE57736 was downloaded from Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/).
Authors’ contributions
XBQ and WJZ participated in the design of this study, and they both performed the statistical analysis. LZ and PJH carried out the study and collected important background information. BJS drafted the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
As the paper did not involve any animals or human experiment, there was no need for ethical approval.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qin, XB., Zhang, WJ., Zou, L. et al. Identification potential biomarkers in pulmonary tuberculosis and latent infection based on bioinformatics analysis. BMC Infect Dis 16, 500 (2016). https://doi.org/10.1186/s12879-016-1822-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-016-1822-6