
Abstract
Background
Variant Creutzfeldt-Jakob Disease (vCJD) is primarily associated with dietary exposure to bovine-spongiform-encephalopathy. Cases may be missed in the elderly population where dementia is common with less frequent referral to specialist neurological services. This study’s twin aims were to determine the feasibility of a method to detect possible missed cases in the elderly population and to identify any such cases.
Methods
A multi-site study was set-up in Lothian in 2016, to determine the feasibility of enhanced CJD-surveillance in the 65 + population-group, and undertake a clinicopathological investigation of patients with features of ‘atypical’ dementia.
Results
Thirty patients are included; 63% male, 37% female. They were referred because of at least one neurological feature regarded as ‘atypical’ (for the common dementing illnesses): cerebellar ataxia, rapid progression, or somato-sensory features. Mean-age at symptom-onset (66 years, range 53–82 years), the time between onset-of-symptoms and referral to the study (7 years, range 1–13 years), and duration-of-illness from onset-of-symptoms until death or the censor-date (9.5 years, range 1.1–17.4 years) were determined. By the censor-date, 9 cases were alive and 21 had died. Neuropathological investigations were performed on 10 cases, confirming: Alzheimer’s disease only (2 cases), mixed Alzheimer’s disease with Lewy bodies (2 cases), mixed Alzheimer’s disease with amyloid angiopathy (1 case), moderate non-amyloid small vessel angiopathy (1 case), a non-specific neurodegenerative disorder (1 case), Parkinson's disease with Lewy body dementia (1 case), and Lewy body dementia (2 cases). No prion disease cases of any type were detected.
Conclusion
The surveillance approach used was well received by the local clinicians and patients, though there were challenges in recruiting sufficient cases; far fewer than expected were identified, referred, and recruited. Further research is required to determine how such difficulties might be overcome. No missed cases of vCJD were found. However, there remains uncertainty whether this is because missed cases are very uncommon or because the study had insufficient power to detect them.
Similar content being viewed by others


Introduction
Prion diseases affect animals and humans and, in humans, exist in idiopathic (sporadic), genetic, and acquired forms. Variant Creutzfeldt-Jakob Disease (vCJD) is a very rare, acquired form of human prion disease, which arose as a zoonosis associated with dietary exposure to bovine spongiform encephalopathy (BSE; a prion disease of cattle). Secondary human to human transmission has occurred via blood (red cell transfusion and factor VIII treatment) [1, 2], but no other transmission routes, such as surgery and dentistry, have been identified [3, 4]. Despite an estimated significant dietary exposure before UK control measures were instituted between 1988 and 1996 [5, 6], only 178 cases of definite or probable vCJD have occurred in the UK with no vCJD deaths reported since 2016 [7]. The fact that relatively few clinical cases have resulted from a potentially large exposure has several possible explanations. Firstly, it is likely that there is a significant species transmission barrier between bovine and man. Secondly, when prion diseases are acquired, there is often a long incubation period (up to 40 years) [8]; in the context of BSE and vCJD, the mean is estimated to be up to 10 years [9, 10]. Susceptibility to disease and incubation period length are related, at least in part, to a polymorphism in the human prion protein gene (PRNP). At PRNP-codon 129, there are 3 common variations resulting in MM, MV, or VV corresponding protein types; about 37% of the UK population is MM, 51% MV and 12% VV [11]. Studies of sporadic (sCJD), kuru and iatrogenic CJD indicate that MM individuals are more susceptible to disease [5, 12,13,14] and, in acquired cases, with a shorter incubation period [15]. This suggests that MM individuals would be expected to be more susceptible to BSE infection and to have the shortest incubation period. All but one of the UK vCJD cases with genetic testing have been of the MM genotype [16]. Overall, further vCJD clinical cases are expected but in uncertain numbers and timing [9]. There is experimental transmision evidence to support the view that MV and VV individuals are less susceptible to vCJD and have longer incubation periods if they develop disease [15]. The same study suggests that some BSE infection might be truly subclinical i.e. infection never resulting in clinical disease. Therefore, there are good grounds for thinking that currently asymptomatic infected individuals exist in the UK population who may be capable of infecting others; a vital public health consideration. Since abnormal prion protein is found in lymphoreticular tissue in vCJD and is present in the preclinical phase, studies have been performed on routine surgical tonsil and appendix samples [17,18,19]. On the basis of these studies, and the assumption that any detected abnormal prion protein reflects BSE infection, the UK population prevalence of asymptomatic vCJD is estimated to be 1 in 2000, although one recent study has suggested that this assumption might be problematic [20]. The estimated prevalence of asymptomatic infection contrasts with the small number of both clinical cases and secondary transmission events. One potential explanation is case under-ascertainment. Arguably under-ascertainment might occur particularly in the elderly, where other commoner dementing brain diseases are common, sCJD (an important differential diagnosis of vCJD) is mainly found, with some evidence that the vCJD clinical phenotype might be different in older patients than in younger ones, autopsy rates are low and specialist neurological referral less frequent [21, 22]. With the overlap in clinical phenotypes of prion diseases, the best ascertainment of vCJD requires ascertainment of all forms of human prion disease, so this study aimed to identify cases of all CJD types. Although the primary study aim was to detect unsuspected cases of vCJD, there was a possible additional outcome of interest. The age-specific mortality rate for sCJD falls off after the age of 80 years [16] whereas current theories of aetiology could argue for a continually increasing incidence with age. Therefore, there is the possibility that there might be underascertainment of elderly sCJD cases especially given generally low autopsy rates [22].
In 2016, a multi-site study was set up in Lothian to determine the feasibility of enhanced CJD surveillance in the 65 + population and, undertake a clinicopathological investigation of patients with atypical features of dementia accessing psycho-geriatric services, thereby, detect otherwise unrecognised prion disease.
A study to ascertain possible under-ascertainment of vCJD cases in the young was based on reporting and reviewing all cases of such deterioration in UK children, however this was possible because the relevant clinical presentation was relatively uncommon [23,24,25]. A similar process would be numerically impossible in the elderly and so some selective criteria (clinical and geographical) were defined, as detailed in the methods section below.
Methods
Base population and sample size
In general, the incidence of neurodegenerative diseases causing dementia increases with age, particularly after the age of 65. Latest UK estimates show around 850,000 people suffer from dementia [26]; the most common diagnoses being Alzheimer’s Disease (AD) and/or vascular dementia (VD), and less frequently, dementia with Lewy bodies (DLB), frontotemporal dementia (FTD), Parkinson’s disease (PD) and other neurodegenerative diseases. The majority of these conditions can be diagnosed with reasonable confidence due to their characteristic presentations and some investigation results, however, around 10% of patients [27] may present atypically with unusual features or lacking characteristic ones, with consequent diagnostic uncertainty; missed cases of prion disease might be found particularly in this group.
In Lothian, 125,000 adults are over 65 years [28]. Of these, around 1% are expected to develop dementia annually [26], of which, around 10% might be considered clinically ‘atypical’. Considering an 80% power for detection of cases with 95% confidence intervals within a 5% margin of error, a sample size of 100 cases/year was deemed appropriate. However, it is difficult to estimate cases that would become known to dementia services out-with primary care and, therefore potentially identified as eligible to join the study.
Eligibility (Table 1)
Cases
Patients aged 65 + with ‘atypical’ features defined as presentations thought, by the referring clinicians, to be different from what they regarded as characteristic for AD, VD, LBD, FTD, and PD, were eligible. New and existing patients with ‘atypical’ features e.g. rapid disease progression, cerebellar ataxia, and somatosensory features, were eligible.
Exclusion: non-prion pathological diagnosis; known inherited non-prion dementia; clear psychiatric diagnosis; space-occupying brain lesion and neuroinflammatory conditions; radiological evidence of a cerebral insult temporally related to symptoms.
Sites
Eligible sites included psychiatry of old age, medicine of the elderly, and neurology (including Ann Rowling Clinic (ARC) specialties, all of which provide dementia services. Sites providing dementia services outside NHS Lothian were excluded.
Recruitment and follow-up
Figure 1 shows the recruitment process. First, an eligible site was identified. Second, a lead clinician was identified as the point of contact who ascertained eligible patients. To protect patient confidentiality, the lead clinician ensured suitability of the patient by checking the patient's records against the study’s eligibility criteria. For those deemed eligible, the clinician briefly discussed the study with the patient if they had capacity, and/or with their representative, and gave them the 65 + study pack: patient study information sheet, consent form and reply slip. For consented patients, the local clinician contacted the study team to countercheck eligibility and referral was made. At the ARC, the lead clinician screened their database to identify eligible patients. Those identified were contacted in writing and the study pack provided. The study nurse arranged a meeting with those who responded.

Recruitment process
All the included cases were followed up initially by telephone within 1 month of recruitment, subsequently, face-to-face every 3 months, and monthly via TraKcare: a comprehensive hospital care management system that records admissions, discharges, and the care provided. From time to time, the clinical research registrar reviewed the cases for evidence of prion disease. Follow-up was until resolution of symptoms or death, but discontinued if a patient/representative withdrew. An email update was sent to the Lothian clinician network forum monthly.
Data collection and investigations
Demographic and referral information was collected. The onset of symptoms was determined as the year of first self-reported clinical symptoms; verified (as much as possible) with the general practitioner’s notes. Referral was noted as the date the case was referred.
Age at onset of symptoms, suspected diagnoses, duration of illness (time between age at onset of symptoms and death or censor date), and past medical, surgical, blood transfusion, family history of CJD and dementia, occupation, and residential histories, were collected.
A combination of specific neurological assessments were done: the Addenbrooke’s Cognitive Examination (ACE-III) [29], the Severe Impairment Battery—Short Form (SIB-S) [30], the MRC Scale [31], a Frontal Assessment Battery (FAB) [32], Hospital Anxiety and Depression Scale (HADS) [33], and the Edinburgh Motor Assessment (EMAS) Scale [34]. Brain Magnetic Resonance Imaging (MRI) scan was offered to all cases as part of their diagnostic work-up, if this had not already been undertaken. For those who had not had recent MRI scans, arrangements were made for a research MRI brain scan at the University of Edinburgh Brain Research Imaging Centre (BRIC), which involved a Diffusion Weighted Imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) (2D or 3D) as part of a standard brain protocol.
If the results evidenced prion disease, the referring clinician was alerted, who then referred the case to the National CJD Research & Surveillance Unit (NCJDRSU). Blood (2 ml) or buccal samples to determine PRNP-129 polymorphism, were collected. None of the cases had a lumbar puncture for Cerebral Spinal Fluid (CSF) tests. These would have been undertaken if indicated by usual clinical criteria/practice, including a strong clinical suspicion of CJD, but they were not part of the research-led investigations.
The possibility of brain tissue donation was raised in life with the patient/representative. In the event of death, where consent was given, a limited, head-only autopsy was arranged. Standard neurological disease histopathology was conducted: sections of tissue from each of the four cortical regions (frontal, temporal, occipital, parietal), the thalamus and the cerebellum were retained,fixed in formalin and processed to paraffin blocks. Each of the six samples was subjected to a standard suite of investigations for neurodegenerative disease using a panel of markers for neurodegenerative proteins (tau, β-amyloid, α-synuclein, p-TDP-43) and screening for spongiform change. Abnormal prion protein was assessed by immunohistochemistry (12F10 and KG9 antibodies) and by biochemistry. Biochemical analysis required the use of approximately 2-3 g of frozen tissue from each of the frontal, temporal, occipital and parietal regions, the thalamus and the cerebellum. Investigations included the most sensitive abnormal prion protein detection methods currently available, which might offer advantages over routine diagnostic techniques in a presumed negative population. These were conducted in series on all samples as follows: standard diagnostic Western blot (WB) for protease-resistant prion protein (PrPres) [35], High sensitivity sodium phosphotungstic acid precipitation(NaPTA) WB for PrPres [36], Conformation dependent immunoassay (CDI) analysis for PrPSc [37], Single round protein misfolding cyclic amplification (PMCA) for ultra-sensitive vCJD PrPSc detection [38] and real-time quaking induced conversion (RT-QuIC) for ultra-sensitive sCJD PrPSc detection [39]. The details of these tests are not included in this paper (but there were no findings suggestive of CJD, including vCJD).
Outcome measures
First, to establish whether the approach used for enhanced CJD surveillance in the 65 + population was feasible, i.e. whether one could recruit suitable patients in sufficient numbers and enhance their assessment, investigation, and diagnosis. Using Bowen’s framework, acceptability, demand, practicality, integration, and efficacy were assessed [40]. Effort expended (staff involved versus numbers recruited), cases expected versus identified, recruitment rate, and proportions consented to clinical-investigations and autopsy were also examined.
Second, to describe the clinicopathological characteristics of the cases to identify whether there were any previously unsuspected prion diseases, and, if so, to determine why they might not be identified in usual clinical practice.
Results
Recruitment occurred between 1st April 2016 and 30th June 2019, with ongoing follow-up to date. Table 2 summarises the characteristics of cases.
Demographics and referral
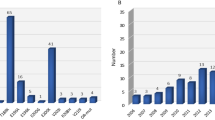
Thirty-one cases were recruited, 1 withdrew, thereby, only 30 cases are considered. The majority were male (63%), and most were below 80 years (84%). Fifty-percent of the referrals came from the ARC. On average, the interval between onset of symptoms and referral was 7 (range 1 – 13) years; with the oldest age group (80 + years) having a relatively short interval on average, of 5 years (range 2 – 10 years). By the censor date (28.02.2021), 70% of the cases had died.
Clinical characteristics
The mean age at onset of symptoms was 66 (range 53 – 82) years. Thirty-seven percent were referred with a suspicion of atypical AD, 30% had unclear/unknown dementia, 20% had mixed dementia, and 13% had FTD. All the cases exhibited at least one ‘atypical’ neurological feature. The mean duration of illness was 9.5 (range 1.1 – 17.4) years.
A range of other clinical symptoms were also noted. All except one case had executive dysfunction (97%). Other common symptoms included: forgetfulness/ memory impairment (93%), language disturbance (expressive (87%) and receptive (60%)), visuospatial difficulties (73%), disturbance in gait (70%), apraxia (77%), slowness of movement (60%) and lack of insight (53%). Psychiatric symptoms were also noted, of which, 80% presented with depression, 70% apathy/ withdrawal, 67% had behavioural disturbances, 63% had anxiety, and 57% had repetitive movements/behaviours. Involuntary movement particurlarly tremor was seen in 60% of the cases. Only 8 cases had sensory symptoms, of which, 5 had numbness/ tingly/ paraesthesia and the remaining 3 had pain/ burning/ discomfort. No case presented with dystonia or akinetic mutism.
Other relevant histories
The majority of cases had at least 1 previous surgical intervention (93%) before referral. Examining the 3-5 year pre-and-post operative period showed no link with cognitive decline. Similarly, no evidence was found between blood products or previous injections (treatments) and cognitive decline. About two-fifths had a family history of dementia, and none, of prion disease. A small proportion worked in professions considered at risk for prion disease, but no evidence of a link was found.
Brain MRI Investigation
Twenty-one cases (70%) consented to new or review of previous brain MRI (Table 3). A wide variation between when the brain MRI was performed and the date of onset of first symptoms was observed (19 days—3,626 days (around 10 months)). All the brain MRIs reviewed were found to have some abnormalities. The most common abnormality identified was atrophy: 6 cases had global/generalised cerebral atrophy (1 case was exaggerated in occipital lobes, 2 cases had moderate atrophy (advanced for age), 1 case also had supatentonial small vessel disease change though static since 2013, and 2 cases were generally noted to have cerebral atrophy advanced for age); 8 cases had focal atrophy/ biparietal volume loss (affecting the temporal, parietal, or occipital lobes) and 1 case was described to have generalised atrophy (unspecified). Microvascular ischaemia/ chronic small vessel disease changes were also noted in 3 cases: 2 cases had moderate microvascular ischaemia, and 1 case, advanced chronic small vessel disease with extensive atrophy in the medial temporal lobes. Two cases had diffuse prominence of the ventriculosulcal system which was advanced for age: 1 case had moderate diffusion and focal atrophy in the left temporal lobe and peri-insular cortex, and the second case had marked diffusion more prominent in temporal and parietal lobes bilaterally. None of the abnormalities identified was suggestive of prion disease, therefore, further clinical investigations were not undertaken.
Genotyping
Eighty-three percent of the cases consented to PRNP-129 genotyping (Table 4): 32% were MM, 52% MV and 16% VV. Carriers of the MM genotype had a shorter duration of illness (8.3 years) than MV (10.4 years) or VV (11.3 years). On average, age at onset of symptoms had minimal variation across the 3 genotypes (MM 64 years, MV 65 years and VV 63 years).
Autopsy neuropathology
Sixty-four percent consented to autopsy neuropathology examination (Table 5). Unfortunately, delays in notification where embalming had taken place and COVID-19 restrictions meant autopsy could not be performed for some cases and was only available for 10 cases. All cases were assessed using validated grading-and-staging systems, and where a neuropathological diagnosis of AD-neuropathological change (AD-NC) was made, a final clinicopathological diagnosis was made based on the current National Institute on Aging-Alzheimer’s Association (NIA-AA) criteria (PMID: 22,265,587). The final pathological diagnosis in the cases examined was: Alzheimer’s disease only (2 cases); mixed Alzheimer’s disease with Lewy body dementia (2 cases); Alzheimer’s disease with severe cerebral amyloid angiopathy (1 case); vascular dementia with non-amyloid small vessel angiopathy (1 case); Lewy body dementia (3 case). No prion disease cases were detected. One case could not be characterised either clinically or pathologically; a rapidly progressive neurodegenerative disorder clinically thought to be frontotemporal dementia although with late onset cerebellar ataxia. No significant pathology was seen with immunohistochemical analysis of tau, β-amyloid, α-synuclein, FUS, p62, polyQ [trinucleotide repeat diosrders], and both 12F10 and KG9 [prion disorders].
Feasibility
The study was well-received, accepted, regarded as appropriate, and perceived to have a positive effect by both patients/representatives and clinicians. The study was seen to provide further clarity on difficult diagnoses. A number of clinicians expressed their intention to refer cases that fitted the study’s criteria. The study was perceived to fit well (to some extent) within the existing dementia services. Despite significant efforts involving several staff (research nurse, clinical research registrar, study neurologist, neuroradiologist, neuropathologist, two post-doctoral laboratory researchers, and an epidemiologist), recruitment fell well below the predicted eligible numbers. It was anticipated that 300 cases might be recruited with only 30 cases recruited (28% of 109 who met the eligibility criteria from 128 potential cases identified and referred). Retention was, however, good with only 1 withdrawal. All the recruited cases underwent clinical examination but only 70% consented to brain MRI investigation or a review of their previous brain MRI investigation report. Only 10 out of the 21 cases that died, underwent autopsy neuropathology examination.
Prion diseases
None detected.
Discussion
This is the first multi-site study, in one UK region (NHS Lothian Health Board), assessing the feasibility of enhanced surveillance of CJD in the 65 + population group to detect otherwise unrecognised prion disease. Thirty cases were recruited exhibiting at least one ‘atypical’ neurological feature. These ‘atypical’ features could suggest prion disease, but, are not unique to prion diseases [41,42,43]. Essentially, a diagnosis of prion disease is based on the clinical pattern, exclusion of other diagnoses, and supportive investigation findings (confirmed by neuropathology). No prion diseases were found.
The PRNP-129 genotyping profile of the study cases (32%—MM, 52%—MV, 16%—VV) reflects a profile of the UK general population (37%—MM, 51% -MV, 12%—VV) [11], but different to sCJD and vCJD [16]. This is not surprising, and while incubation and susceptibility to prion disease have been linked to PRNP-codon-129 polymorphism, it does not help to distinguish prion from non-prion diseases, and broadly, its role in other neurodegenerative diseases is inconclusive [44, 45]. The duration of illness of the cases was widely varied from 1.1 years to 17.4 years. In itself, the long duration of illness makes a diagnosis of vCJD (median duration, 14 months) or sCJD (median duration, 4 months) improbable [46, 47]. However, unusually long duration in prion disease is recognised [5]. The time interval between symptom onset and referral ranged from 1 to 13 years being shorter on average for the oldest age group (80 +) compared with younger cases. Arguably, this might indicate the possibility that prion disease in the younger 65 + population might have been missed. Another possibility is that a smaller proportion of the younger age range, relative to the older range, might be known to dementia services out-with primary care. However, it is unclear why there was a small number of cases referred after the age of 80 years where the mortality rate from sCJD also declines [16].
The study was, primarily, a feasibility study, trying to determine if it was a successful method of enhanced CJD surveillance in the elderly. While the approach used was well received, there are obvious problems. Firstly, actual numbers recruited did not match predicted numbers due partly to fewer than expected referrals (which does not rule out the possibility of an error in the predicted calculations) and a relatively low participation rate. Secondly, the study relied on the ability of local clinicians to refer eligible cases, who, may not have been able to identify the right cases for various reasons, and if they did, the patients/representative may not have wanted to be referred. Low referral could also have been due to the local clinicians' significant workloads, therefore, no time to speak about the study with potential cases. All these factors are not uncommon in research studies [48,49,50]. Thirdly, participation was limited both in joining the study (85% referred were eligible) and in full consent (70% agreeing to MRI and 73% with autopsy consent, of which, 8 are alive). Full participation was likely to have been limited by the patient’s/representative’s perception of the study. Precise reasons are unclear, however, perceptions of risks vs. benefits [51] and fear may have had some influence.
The limited recruitment number required the efforts of several study personnel and it is difficult to see how an extension to a larger population would be feasible. The study did not find any missed cases of prion disease, but, given the numbers and methodology involved, it is difficult to draw any firm conclusions on whether there are undetected cases in the elderly. Neuropathology is the final diagnostic determinant but this was available in only 10 cases. An additional 4 cases were missed, which was an unfortunate consequence both due to delays in alerting the study team therefore embalming took place, and the unexpected COVID-19 crisis.
Conclusion
The results suggest the methodology used is not feasible as a basis for more extended enhanced surveillance for prion disease in the elderly. However, no previously unsuspected cases of prion disease were identified. There were lower than expected referral rates and incomplete recruitment. A review investigating these aspects is being undertaken.
Availability of data and materials
The data is held by the NCJDRSU as part of their surveillance projects. The NCJDRSU is an internationally recognised World Health Organisation reference centre and European Centre for Disease Control hub for diagnosis of all forms of human prion disease and has substantial expertise in prion disease surveillance and clinical and laboratory research in neurology, neuropathology, brain imaging and biochemical investigations in relation to dementing illness (www.cjd.ed.ac.uk). Enquiries to access the data can be made to NCJDRSU (https://www.cjd.ed.ac.uk/contact-us), which will be considered on a case by case basis in line with the NCJDRSU Data Protection and Security Code of Practice.
Abbreviations
- ACE:
-
Addenbrooke’s Cognitive Examination
- AD:
-
Alzheimer’s Disease
- AD-NC:
-
Alzheimer’s Disease—Neuropathological change
- ARC:
-
Ann Rowling Clinic
- BSE:
-
Bovine Spongiform Encephalopathy
- CDI:
-
Conformation Dependent Immunoassay
- CJD:
-
Creutzfeldt-Jakob Disease
- CSF:
-
Cerebral Spinal Fluid
- DLB:
-
Dementia with Lewy Bodies
- DWI:
-
Diffusion Weighted Imaging
- EMAS:
-
Edinburgh Motor Assessment
- FAB:
-
Frontal Assessment Battery
- FLAIR:
-
Fluid-Attenuated Inversion Recovery
- FTD:
-
Frontotemporal Dementia
- HADS:
-
Hospital Anxiety and Depression
- MRC:
-
Medical Research Council prion disease rating scale
- MRI:
-
Magnetic Resonance Imaging
- NaPTA:
-
High sensitivity sodium phosphotungstic acid precipitation
- NHS:
-
National Health Service
- NIA-AA:
-
National Institute on Aging-Alzheimer’s Association
- PD:
-
Parkinson’s Disease
- PMCA:
-
Protein Misfolding Cyclic Amplification
- PRNP:
-
Human Prion Protein Gene
- RT-QuIC:
-
Real-time quaking induced conversion
- sCJD:
-
Sporadic Creutzfeldt-Jakob Disease
- SIB-S:
-
Severe Impairment Battery—Short Form
- UK:
-
United Kingdom
- vCJD:
-
Variant Creutzfeldt-Jakob Disease
- VD:
-
Vascular Dementia
- WB:
-
Western blot
References
Urwin PJ, Thanigaikumar K, Ironside JW, Molesworth A, Knight RS, Hewitt PE, Llewelyn C, Mackenzie J, Will RG. Sporadic Creutzfeldt-Jakob disease in 2 plasma product recipients, United Kingdom. Synopsis. 2017;23:6.
Urwin PJ, Mackenzie JM, Llewelyn CA, Will RG. Creutzfeldt-Jakob disease and blood transfusion: updated results of the UK Transfusion Medicine Epidemiology Review Study. Vox Sang. 2006;110:310–6.
Collins S, Law MG, Fletcher A, Boyd D, Kaldor J, Masters CL. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: a case-control study. Lancet. 1999;353(9154):P693-697.
Everington D, Smith AJ, Ward HJT, Letters S, Will RG, Bagg J. Dental treatment and risk of variant CJD - a case control study. Br Dent J. 2007;202(8):470–1.
Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull. 2003;66:255–65.
National Institute for Health and Care Excellence. Reducing the risk of transmission of Creutzfeldt-Jakob Disease (CJD) from surgical instruments for interventional procedures on high risk tissues. Accessed 26 Mar 2021. Available at: 2 Indication | Reducing the risk of transmission of Creutzfeldt–Jakob disease (CJD) from surgical instruments used for interventional procedures on high-risk tissues | Guidance | NICE.
National CJD Research & Surveillance Unit. Latest NCJDRSU CJD monthly statistics. Available at: Data and Reports | CJD (ed.ac.uk).
Department of Health and Social Care. Guidance. Minimise transmission risk of CJD and vCJD in healthcare settings. Prevention of CJD and vCJD by the Advisory Committee on Dangerous Pathogens’ Transmissible Spongiform Encephalopathy (ACDP TSE) subgroup. Accessed 26 Mar 2021. Available at: ~4175378.doc (publishing.service.gov.uk).
Ghani A, Donnelly C, Ferguson N, Anderson R. Biologies. 2002;325:37–41.
European Centre for Disease Control. Facts about variant Creufeldt-Jakob Disease. Accessed 25 Mar 2021. Available at: Facts about variant Creutzfeldt-Jakob disease (europa.eu).
Mackay G, Knight R, Ironside J. The molecular epidemiology of variant CJD. Int J Mol Epidemiol Genet. 2011;2(3):217–27.
Collinge J, Palmer MS, Dry den AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991;337:1441–2.
Hauw JJ, Sazdovitch V, Laplanche JL, Peoc’h K, Kopp N, Kemeny J, et al. Neuropathologic variants of sporadic Creutzfeldt-Jakob disease and codon 129 of PrP gene. Neurology. 2000;54:1641–6.
Douet J, Huor A, Cassard H, Lugan S, Aron N, Mesic C, Vilette D, Barrio T, Streichenberger N, Perret-liaudet A, Delisle M, Péran P, Deslys J, Comoy E, Vilotte J, Goudarzi K, Béringue V, Barria MA, Ritchie DL, Ironside JW, Andréoletti O. Prion strains associated with iatrogenic CJD in French and UK human growth hormone recipients. Acta Neuropathol Commun. 2021;9:1.
Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, Tuzi NL, Head MW, Ironside JW, Will RG, Manson JC. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5(5):393–8.
Creutzfeldt-Jakob Disease Surveillance in the UK. 28th Annual Report 2019. Available at: https://www.cjd.ed.ac.uk/surveillance
Clewley JP, Kelly CM, Andrews N, et al. Prevalence of disease related prion protein in anonymous tonsil specimens in Britain: cross sectional opportunistic survey. BMJ. 2009;338:b1442.
Gill ON, Spencer Y, Richard-Loendt A, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013;347:f5675.
Hilton DA, Ghani AC, Conyers L, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203(3):733–9.
Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Brown D, Sinka K, Andrews N, Dabaghian R, Simmons M, Edwards P, Bellerby P, Everest DJ, McCall M, McCardle LM, Linehan J, Mead S, Hilton DA, Ironside JW, Brandner S. Prevalence in Britain of abnormal prion protein in human appendices before and after exposure to the cattle BSE epizootic. Acta Neuropathol. 2020;139:965–76.
El Tawil S, Mackay G, Davidson L, Summers D, Knight R, Will R . Variant Creutzfeldt-Jakob disease in older patients. J Neurology Neurosurg Psychiatry. 2015. https://doi.org/10.1136/jnnp-2014-309397.NP 2015 Jan 21. pii: jnnp-2014–309397.
Turnbull A, Osborn M, Nicholas N. Hospital autopsy: Endangered or extinct? J Clin Pathol. 2015;68(8):601–4 PMID: 26076965.
Verity CM, Nicoll A, Will RG, Devereux G, Stellitano L. Variant Creutzfeldt-Jakob disease in UK children: a national surveillance study. Lancet. 2000;356:1224–7.
Devereux G, Stellitano L, Verity CM, Nicoll A, Will RG, Rogers P. Variations in neurodegenerative disease across the UK: findings from the national study of Progressive Intellectual and Neurological Deterioration (PIND). Arch Dis Child. 2004;89:8–12.
Verity C, Winstone AM, Stellitano L, Will R, Nicoll A. The epidemiology of progressive intellectual and neurological deterioration in childhood. Arch Dis Child. 2010;95:361–4 (deterioration in childhood. Arch Dis Child 2010; 95:361-364).
Alzheimer’s Society. Dementia UK. 2nd edition. 2014. http://www.alzheimers.org.uk/site/scripts/documents_info.php?documentID=2759
National CJD Research & Surveillance Unit. The 65+ dementia study protocol version 4.0. 2020. Available at: The 65+ Dementia Study: Enhanced surveillance of Creutzfeldt-Jakob Disease in the older population | CJD.
National Records of Scotland. 2011 Census Reconciliation Report - Population. Available at: 2011 Census Reconciliation Report - Population | National Records of Scotland (nrscotland.gov.uk).
Mioshi E, Dawson K, Mitchell J, Arnold R, Hodges JR. The Addenbrooke’s Cognitive Examintion Revised (ACE-R): A brief cognitive battery test for dementia screening. Int J Geriatr Psychiatry. 2006;21:1078–85.
Saxton J, Kastango KB, Hugonot-Diener L, Boller F, Verny M, Sarles CE, Girgis RR, Devouche E, Mecocci P, Pollock BG, DeKosky ST. Development of a short form of the Severe Impairment Battery. Am J Geriatr Psychiatry. 2005;13(11):999–1005.
Thompson AGB, Lowe J, Fox Z, Lukic A, Porter M, Ford L, Gorham M, Gopalakrishnan GS, Rudge P, Walker AS, Collinge J, Mead S. The Medical Research Council Prion Disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013;136:1116–27.
Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at the beside. Neurology. 2000;55:1621–6.
Zigmond AS, Snaith RP. The Hospital Anxiety and depression scale. Acta Psychiatr Scand. 1983;67:361–70.
Bak TH. Edinburgh Motor Assessment (EMAS). 2013. Accessed 20 Sept 2019. Available at: https://www.era.lib.ed.ac.uk/bitstream/handle/1842/8225/EMAS%201%20Dec%202013.pdf;jsessionid=8929B5D6519981336AD4C3443812907A?sequence=1
Head MW, Yull HM, Ritchie DL, Langeveld JP, Fletcher NA, Knight RS, Ironside JW. Variably protease-sensitive prionopathy in the UK: a retrospective review 1991–2008. Brain. 2013;136(4):1102–15.
Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW. Peden Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia. 2010;16(2):296–304. https://doi.org/10.1111/j.1365-2516.2009.02181.x (Epub 2010 Jan 12).
Peden AH, Sarode DP, Mulholland CR, Barria MA, Ritchie DL, Ironside JW, Head MW. The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol Commun. 2014;2:152. https://doi.org/10.1186/s40478-014-0152-4.
Barria MA, Balachandran A, Morita M, Kitamoto T, Barron R, Manson J, Knight R, Ironside JW, Head MW. Molecular barriers to zoonotic transmission of prions. Emerg Infect Dis. 2014;20(1):88–97. https://doi.org/10.3201/eid2001.130858.
Peden AH, McGuire LI, Appleford NE, Mallinson G, Wilham JM, Orrú CD, Caughey B, Ironside JW, Knight RS, Will RG, Green AJ, Head MW. Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J Gen Virol. 2012;93(Pt2):438–49. https://doi.org/10.1099/vir.0.033365-0. Epub 2011 Oct 26.
Bowen DJ, Kreuter M, Spring B, Cofta-Woerpel L, Linnan L, Weiner D, Bakken B, Kaplan CP, Squiers L, Fabrizio C, Fernandez M. How we design feasibility studies. Am J Prev Med. 2009;36(5):452–7.
Geschwind M, Shu H, Haman A. Sejvar, JJ, and Miller, BL. Rapid progressive dementia. Ann Neurol. 2008;64(1):97–108.
Mead S, Rudge P. CJD Mimics and Chameleons. P Pract Neurol. 2017;17:113–21.
Schmidt C, Redyk K, Meissner B, et al. Clinical features of rapidly progressive Alzheimer’s disease. Dement Geriatr Cogn Disord. 2010;29:371–8.
Combarros O, Sanchez-Guerra M, Llorca J, Alvarez-Arcaya A, Berciano J, Pena N, et al. Polymorphism at codon 129 of the prion protein gene is not associated with sporadic AD. Neurology. 2000;55:593–5.
Karamujic-Comic H, Ahmad S, LysenTS, Heshmatollah A, Roshchupkin GV, Vernooij MW, Rozemuller AJM, Ikram MA, Amin N, van Duijn CM. Prion protein codon 129 polymorphism in mild cognitive impairment and dementia: the Rotterdam study. Brain Commun. 2020;2(1):1–7. https://doi.org/10.1093/braincomms/fcaa030.
Brandel JP, Knight R. Variant Creutzfeldt-Jakob Disease. Chapter 11 in Handbook of Clinical Neurology,Vol. 153 (3rd series). Human Prion Diseases M. Pocchiari and J. Manson, Editors. https://doi.org/10.1016/B978-0-444-63945-5.00011-8
Will RG, Ironside JW. Sporadic and Infectious Human prion diseases. Cold Spring Harb Perspect Med. 2017;7:a024364.
Thoma A, Farrokhyar F, McKnight L, Bhandari M. Practical tips for surgical research: how to optimize patient recruitment. Can J Surg. 2010;53(3):205–10.
Donovan J, Parmasivan S, de Salis I, Torrien M. Clear obstacles and hidden challenges: understanding recruiter perspectives in six pragmatic randomised controlled trials. Trials. 2014;15(1):5.
Hamilton M, Genge A, Johnston M, Lam D, Mobach T, Marriott J, et al. Patient recruitment by neurological registries. Can J Neurol Sci. 2013;40(Suppl 2):S23–6.
Braunstein JB, Sherber NS, Schulman SP, Ding EL, Powe NR. Race, medical researcher distrust, perceived harm, and willingness to participate in cardiovascular prevention trials. Medicine. 2008;87(1):1–9. https://doi.org/10.1097/md.0b013e3181625d78.
Acknowledgements
Our sincere thanks and appreciation go to Tracy Millar and Chris Lerperniere for their help in coordinating authorisations of brain tissue donations and post-mortem investigations.
Funding
This work is independent research commissioned and funded by the Department of Health and Social Care Policy Research Programme (PR-ST-1214–10002). The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Author information
Authors and Affiliations
Contributions
RK, AM, and CS conceptualised the study. GL, BW, and LK collected the data. LK analysed the data and drafted the manuscript with inputs from RK. All authors reviewed the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by Scotland A Research Ethics Committee (ref:15/SS/0196). The cases were recruited only after providing informed consent themselves and/or their legal representatives. The NCJDRSU is an internationally recognised World Health Organisation reference centre and European Centre for Disease Control hub for diagnosis of all forms of human prion disease and is governed by relevant guidelines and regulations, which the study followed.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kanguru, L., Logan, G., Waddel, B. et al. A clinicopathological study of selected cognitive impairment cases in Lothian, Scotland: enhanced CJD surveillance in the 65 + population group. BMC Geriatr 22, 603 (2022). https://doi.org/10.1186/s12877-022-03280-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12877-022-03280-4