
Abstract
Background
Growing evidence has suggested that gut microbiota is closely related to the risk of irritable bowel syndrome (IBS), but whether there is a causal effect remains unknown. We adopted a Mendelian randomization (MR) approach to evaluate the potential causal relationships between gut microbiota and the risk of IBS.
Methods
Genetic instrumental variables for gut microbiota were identified from a genome-wide association study (GWAS) of 18,340 participants. Summary statistics of IBS were drawn from a GWAS including 53,400 cases and 433,201 controls. We used the inverse-variance weighted (IVW) method as the primary analysis. To test the robustness of our results, we further performed the weighted-median method, MR-Egger regression, and MR pleiotropy residual sum and outlier test. Finally, reverse MR analysis was performed to evaluate the possibility of reverse causation.
Results
We identified suggestive associations between three bacterial traits and the risk of IBS (odds ratio (OR): 1.08; 95% confidence interval (CI): 1.02, 1.15; p = 0.011 for phylum Actinobacteria; OR: 0.95; 95% CI: 0.91, 1.00; p = 0.030 for genus Eisenbergiella and OR: 1.10; 95% CI: 1.03, 1.18; p = 0.005 for genus Flavonifractor). The results of sensitivity analyses for these bacterial traits were consistent. We did not find statistically significant associations between IBS and these three bacterial traits in the reverse MR analysis.
Conclusions
Our systematic analyses provide evidence to support a potential causal relationship between several gut microbiota taxa and the risk of IBS. More studies are required to show how the gut microbiota affects the development of IBS.
Similar content being viewed by others


Background
Irritable bowel syndrome (IBS) is a chronic functional gastrointestinal disorder that affects 11% of the world’s population [1]. IBS affects more women than men, and adults younger than 50 years of age compared with older ones [2]. The main symptoms of IBS include abdominal pain, changes in defecation habits and/or fecal condition, abdominal distension, and discomfort [3]. IBS imposes a large burden on patients, impairing health-related quality of life and work productively [4]. Traditional therapeutic approaches for IBS, including dietary changes and antibiotic therapy, may not obtain satisfactory outcomes since most of them are treating symptoms. Recently, the prevalence of IBS has been rising all over the world, mainly due to anxiety and stress [5].
The pathophysiological mechanisms underlying IBS are multifactorial and have been poorly understood. A heritable component of IBS is long recognized in family and twin studies [6]. Evidence is now accumulating that genetic risk in IBS spans from complex polygenic conditions with combinations of common variants to cases with rare single gene abnormalities [7, 8]. Recent studies have shown that gut microbiota may be related to the pathogenesis of IBS [9,10,11]. Treatment with antibiotics or fecal microbiota transplantation relieves global IBS symptoms without causing constipation, suggesting a direct relationship between gut microbiota and IBS [12, 13]. A recent systematic review has pointed out that alterations of gut microbiota exist in patients with IBS, which might exert a pivotal role in the development of IBS [14].
Although gut microbiota has been related with IBS, the causal nature is elusive. Mendelian randomization (MR) analysis is a statistical approach that aims to infer potentially causal relationships from observational association results [15]. MR uses genetic variants associated with exposure as a surrogate for exposure to assess the relationship between the surrogate and the outcome [16]. In recent years, MR analysis has been applied to assess the potential causal relationships between gut microbiota and disease-risking genes [17,18,19]. So far, there is an urgent need to investigate the potential causal relationship between gut microbiota and the risk of IBS.
In the present study, in order to explore the potential causal relationship between gut microbiota and IBS, and to identify specific pathogenic bacteria taxa, we conducted a two-sample MR study based on genome-wide association study (GWAS) summary data.
Methods
Outcome data sources
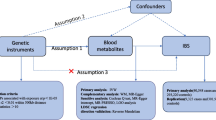
The overall design of the present study is presented in Fig. 1. Briefly, genetic summary statistics for IBS were generated from a GWAS including 53,400 cases and 433,201 controls of European ancestry, which combined data from UK Biobank and Bellygenes initiative [20]. All patients with IBS satisfied at least one of the following four conditions: 1) satisfied the Rome III symptom criteria for IBS diagnosis and did not have other explanations for their symptoms; 2) they admitted that they have been diagnosed with IBS; 3) they self-reported they met IBS diagnosis; and 4) linked hospital episode statistics indicating inpatient or day-case admission with clinician diagnosis of IBS entered as ICD-10 diagnosis [20].

The study design of the associations of gut microbiota and irritable bowel syndrome. Abbreviations: MR, Mendelian randomization; SNP, single nucleotide polymorphism
The summary statistics for human gut microbiome we used in this study were obtained from the most recent GWAS meta-analysis, which included 18,340 participants from 24 cohorts [21]. Detailed of the study has been described elsewhere [21]. Briefly, the study coordinated 16S rRNA gene sequencing profiles and genotyping data from cohorts from the USA, Canada, Germany, Denmark, the Netherlands, Belgium, Sweden, Finland, the UK and so on, and performed the association analyses with adjustment for age, sex, technical covariates, and genetic principal components [21]. As the present study was based on public summary data, no additional ethics approval or consent to participate was required. The details of the data sources in this MR study are shown in Table 1.
Selection of instrumental variables
We first removed 15 bacterial traits without specific name, leaving 196 bacterial traits, including 9 Phylum, 16 Class, 20 Order, 32 Family and 119 Genus. Then, we selected the instrumental variables (IVs) at p < 1.0 × 10–5. In order to obtain IVs from independent loci, we set the linkage disequilibrium (LD) threshold at R2 < 0.001 and clumping distance = 10,000 kb in 1000 Genomes EUR data using “TwoSampleMR” packages. Single nucleotide polymorphisms (SNPs) with the lowest p-value for the associated trait were retained for clumping with 196 bacterial traits. A total of 2699 independent SNPs were found to be associated with 196 bacterial traits. In the reverse MR analysis, we selected IVs associated with IBS at a stricter threshold (p < 5 × 10–8) which has been described in the previous study (Table 2) [20]. After extracted relevant information such as effect allele, effect size including β-value, standard error and P-value for each SNP, we calculated the proportion of variation explained (R2) and F-statistics to quantify the instrument strength, with the following equation: R2 = 2 × MAF × (1 − MAF) × β2, F = R2 (n-k-1) / k(1-R2), where "MAF" is the minor allele frequency of SNPs used as IVs, "n" is the sample size, and "k" is the number of IVs employed [22, 23].
Statistical analysis
We used several methods to estimate the potential causal relationships between gut microbiota and IBS, including fixed/random-effects inverse-variance weighted (IVW) method, weighted median method, MR-Egger regression and MR pleiotropy residual sum and outlier (MR-PRESSO) test. We used the IVW method as the main analysis because it provides the most precise effect estimates and almost all MR-analysis used it as the main analysis [24,25,26]. The IVW method first calculated the ratio estimates for individual SNPs by using the Wald estimator and Delta method, and then combined the estimates which have been calculated from each SNP, thus obtaining the primary causal estimate [27]. Cochran’s Q test was used to test the heterogeneity among the SNPs we selected, and the random-effects IVW method was chosen if heterogeneity exists (p < 0.05) or else fixed-effects IVW method was used [28]. Since the result of IVW method is susceptible to the influences of valid instruments and potential pleiotropic effects, we performed sensitivity analyses to assess the robustness of the association. First, we used the weighted median method to estimate associations since it could provide more reliable estimates of a causal effect when lacking valid instruments [29]. It could provide valid causal effect estimates when less than 50% of information comes from invalid instruments [29]. Second, MR-Egger regression was used to test the potential horizontal pleiotropy, and if the p-value of the intercept was less than 0.05, horizontal pleiotropy of SNPs might exist [30]. Finally, we performed the MR-PRESSO test which conducted a global test of heterogeneity to identify if the SNPs existed possible outliers and obtain a corrected association result after removing the potential outliers [31].
To further assess the influence of potential directional pleiotropy, we scanned each of the SNPs used as IVs for their potential secondary phenotypes using the GWAS Catalog (http://www.ebi.ac.uk/gwas, last accessed on November 22, 2022) and performed MR analyses again after excluding the SNPs associated with other phenotypes.
The associations between human gut microbiota and the risk of IBS were presented as odds ratios (ORs) with their 95% confidence intervals (CIs). We corrected for multiple comparisons using the Bonferroni approach at different taxonomic rank and set statistical significance at a different p-value (p-value < 5.6 × 10–3 for Phylum, p-value < 3.1 × 10–3 for Class, p-value < 2.5 × 10–3 for Order, p-value < 1.6 × 10–3 for Family and p-value < 4.2 × 10–4 for Genus) based on the number of bacterial traits in the specific gut microbiota rank. If a p-value was between the significance threshold and 0.05, we considered suggestive evidence for a potential causal association [25]. Only if all MR methods support the association between the gut microbiota and IBS, the reverse MR analysis was performed. All MR analyses were performed using R version 3.6.3 (https://www.r-project.org/) with “Mendelian Randomization”, “TwoSampleMR” and “MR-PRESSO” packages.
Results
Main results of the 196 bacterial traits with the risk of IBS
The F-statistics for the 196 bacterial traits were ranged from 21.63 to 144.84, which were all above 10, suggesting less possibility to suffer from weak instrument bias. As for the variances of these 196 bacterial traits explained by the IVs, it was estimated to be ranged from 0.57% to 10.11%. The MR results of the associations between all 196 bacterial traits and the risk of IBS are presented in Additional file 1: Table S1. Briefly, we observed suggestive evidence for 11 bacterial traits to be associated with the risk of IBS using IVW method (Fig. 2). The information of IVs used for these 11 bacterial traits are listed in Additional file 1: Table S2.

Forest plot of the associations between genetically determined 11 bacterial traits with the risk of irritable bowel syndrome. Abbreviations: CI, confidence interval; OR, odds ratio; SNP, single nucleotide polymorphism
In particular, we found that genetically predicted phylum Actinobacteria were positively correlated with the risk of IBS [odds ratio (OR): 1.08; 95% confidence interval (CI): 1.02, 1.15; p = 0.011] in the IVW method (Fig. 3). The association between phylum Actinobacteria and IBS remained stable in the weighted-median method (OR: 1.10; 95% CI: 1.01, 1.21; p = 0.030). Furthermore, the MR-PRESSO test did not detect any outliers and the results were similar with the primary method (OR: 1.08; 95% CI: 1.00, 1.17; p = 0.049). In the MR-Egger regression, there was no evidence of directional pleiotropic effects (intercept p-value = 0.270).

Scatter plot of the associations of genetic variants with three bacterial traits and the risk of irritable bowel syndrome. Abbreviations: IBS, irritable bowel syndrome; MR, mendelian randomization; SNP, single nucleotide polymorphism
As for genus Flavonifractor, it was also positively associated with the risk of IBS in IVW method (OR: 1.10; 95% CI: 1.03, 1.18; p = 0.005) (Fig. 3). The results from the weighted-median method were consistent (OR: 1.13; 95% CI: 1.03, 1.24; p = 0.001). The finding of MR-PRESSO test also supported this result (OR: 1.10; 95% CI: 1.04, 1.16; p = 0.008). Intercept of MR-Egger regression also showed no potential horizontal pleiotropy (intercept p-value = 0.252).
In contrast, genus Eisenbergiella was negatively associated with IBS risk using IVW method (OR: 0.95; 95% CI: 0.91, 1.00; p = 0.030) (Fig. 3). In sensitivity analyses, the weighted median method produced similar estimates (OR: 0.92; 95% CI = 0.87, 0.98; p = 0.007), though with wider confidence intervals. Additionally, little evidence of directional pleiotropy was found in MR-Egger regression (intercept p-value = 0.071) and no outliers were detected with the MR-PRESSO test and the effect estimate was similar (OR: 0.95; 95% CI: 0.91, 0.99; p = 0.037).
In addition, we noticed that the rest of eight bacterial traits were suggestively associated with a higher risk of IBS in IVW method (OR: 1.05; 95% CI: 1.01, 1.10; p = 0.023 for class Melaibacteria; OR: 1.06; 95% CI: 1.02, 1.11, p = 0.008 for order Gastraerophilales; OR: 1.06; 95% CI: 1.01, 1.11; p = 0.028 for order Rhodospirillales; OR: 1.07; 95% CI: 1.01, 1.13; p = 0.025 for family Rikenellaceae; OR: 1.08; 95% CI: 1.02, 1.15; p = 0.011 for genus Eubacterium hallii group; OR: 1.07; 95% CI: 1.00, 1.14; p = 0.039 for genus Coprococcus 1; OR: 1.06; 95% CI: 1.02, 1.11, p = 0.006 for genus Prevotella 9; OR: 1.06; 95% CI: 1.00, 1.12; p = 0.046 for genus Ruminiclostridium 6), but results from the weighted median method did not support such a causal effect.
To further assess the influence of potential directional pleiotropy on the causal effect estimates, we used the GWAS Catalog to scan the SNPs associated with these 11 bacterial traits and only four SNPs were found to be accompanied with other traits (Table 3). After excluding these pleiotropic SNPs, we recalculated the F-statistics for the updated IV sets, and the associations of phylum Actinobacteria, genus Eubacterium hallii group and Flavonifractor with the risk of IBS remained stable in the IVW method (OR: 1.08; 95% CI: 1.01, 1.15; p = 0.017 for phylum Actinobacteria, F-statistics = 24.18; OR: 1.07; 95% CI: 1.01, 1.14; p = 0.021 for genus Eubacterium hallii group, F-statistics = 34.11; OR: 1.10; 95% CI: 1.03, 1.19; p = 0.007 for genus Flavonifractor, F-statistics = 41.76). However, the relationship between genus Ruminiclostridium 6 and IBS was unstable (OR: 1.05; 95% CI: 0.99, 1.11; p = 0.081, F-statistics = 37.72).
The result of reverse MR analysis
Finally, we evaluated the potential reverse associations of three bacterial traits and IBS using the reverse MR analyses. We did not find statistically significant associations between IBS and any of these three bacterial traits using IVW method (OR: 1.04; 95% CI: 0.83, 1.31; p = 0.692 for phylum Actinobacteria; OR: 0.80; 95% CI: 0.53, 1.21; p = 0.290 for genus Eisenbergiella and OR: 1.00; 95% CI: 0.74, 1.34; p = 0.980 for genus Flavonifractor). The results were stable across sensitivity analyses, which are listed in Table 4.
Discussion
This two-sample MR study identified a total of 11 bacterial taxa, including phylum Actinobacteria, class Melaibacteria, order Gastraerophilales and Rhodospirillales, family Rikenellaceae, and genus Eubacterium hallii group, Eisenbergiella, Flavonifractor, Coprococcus 1, Prevotella 9 and Ruminiclostridium 6, might be associated with the risk of IBS. However, sensitivity analyses using different MR methods and restricted IV sets demonstrated three bacterial taxa, Actinobacteria, Flavonifractor, and Eisenbergiella, were associated with the risk of IBS.
Phylum Actinobacteria, one of the major phyla of gut microbiota, is pivotal in the maintenance of gut homeostasis [32]. Disorder of Actinobacteria was associated with several diseases, including inflammatory bowel disease [33], ankylosing spondylitis [34], and type 2 diabetes [35]. A decrease of Actinobacteria was found in patients with IBS compared to healthy controls [36]. The reason might be that Actinobacteria as the initial factor of IBS, the host could produce specific antibodies to reduce the abundance of Actinobacteria after IBS occurring. In addition, the abundance of Actinobacteria showed significant alterations after treatment of IBS [37, 38]. The potential causal relationship between Actinobacteria and IBS observed in this study once again suggested the importance of Actinobacteria in the development of IBS.
Genus Flavonifractor, a flavonoid degrader, has also been identified as a risk factor of IBS. The flavonoid compound could alleviate intestinal inflammation of IBS via macrophage-intrinsic AhR [39]. Genus Flavonifractor and its species Flavonifractor plautii were enriched in the stool communities in children with IBS [40]. In addition, Flavonifractor plautii was correlated with recurrent abdominal pain and could elicit enhanced IgG responses in postinfectious IBS patients [41]. Enrichment of the genus Flavonifractor was described in adults with comorbid IBS diarrhea-predominant and depression [42]. A previous study also suggested that dietary modifications could decrease the abundance of Flavonifractor to reduce abdominal pain or accelerated transit time in IBS [43]. Taken together, these studies suggested that a high level of Genus Flavonifractor may be positively associated with the risk of IBS, which is consistent with our findings.
Genus Eisenbergiella was the only identified bacterial taxa being negatively associated with the risk of IBS in this study. However, there was no study reporting the alteration of genus Eisenbergiella in IBS patients to date. In animal studies, only one literature reported that genus Eisenbergiella showed an increasing trend in the IBS group compared to the control group [44]. Even so, genus Eisenbergiella was probably related to eubiosis because it could produce butyrate, acetate, lactate, and succinate as major metabolic products, with a trophic effect on the mucosa [45]. Besides, genus Eisenbergiella might be closely related to the reduction in intestinal inflammation in ulcerative colitis mice [46]. Although this study firstly showed a potential causal relationship between genus Eisenbergiella and the risk of IBS, further research is needed to explore the underlying biological mechanism between them.
Many previous studies showed that patients with IBS were usually accompanied by gut microbiota dysbiosis, but they were observational studies [9, 47]. This study strengthened the causal effects of gut microbiota on IBS by using a genetic epidemiological approach. In addition, the F-statistic of IVs we used all satisfied the threshold of > 10 which suggested that our analyses were less likely to suffer from weak instrument bias. We further performed a reverse MR analysis that excluded reverse causality. Causal association research will be the future direction of studying the role of gut microbiota in the development of diseases. Nowadays, there were many kinds of research focusing on the role of certain gut bacteria in the disease development using animal models [48, 49]. Our MR analysis results may provide a guide for selecting individual gut bacteria to study the role of gut microbiota in the pathogenesis of IBS.
Nevertheless, our study had several limitations. First, bacterial taxa were only analyzed at the genus level but not at a more specialized level such as species or strain levels. Second, while the majority of the participants enrolled in this GWAS are of European descent, the inclusion of participants with other ethnicities may influence the results. Consequently, the generalization of our findings to other racial groups may be subject to limitations. Third, we selected the IVs for gut microbiota at p < 1.0 × 10−5 which were larger than traditional genome-wide significance level (p < 5 × 10–8) to obtain sufficient IVs. In addition, the effect of the bacterial traits we reported was relatively weak and there was no other independent GWAS of IBS with sufficient sample size to validate our findings. Finally, since information of IBS subtypes were not available, further studies are warranted when this information become available.
Conclusions
In conclusion, this study assessed the potential causal role of gut microbiota on the risk of IBS, and found three bacterial taxa, phylum Actinobacteria, genus Flavonifractor and Eisenbergiella may have a suggestive causal relationship with the risk of IBS, which may provide clues for the pathogenesis and novel treatment of IBS.
Availability of data and materials
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number (PRJNA673102, PRJNA683912, PRJEB14839, EGAS00001004420, PRJEB14839, ERP117287, R000635, EGAS00001001704, EGAS0000100924, SRP097785, ERP016332, PRJEB11532, EGAS00001004869, and ERP015317) can be found in the article [21] or Supplementary Material.
Abbreviations
- CI:
-
Confidence interval
- GWAS:
-
Genome-wide association study
- IBS:
-
Irritable bowel syndrome
- ICD:
-
International classification of diseases
- IV:
-
Instrumental variable
- IVW:
-
Inverse-variance weighted
- LD:
-
Linkage disequilibrium
- MR:
-
Mendelian randomization
- MR-PRESSO:
-
MR pleiotropy residual sum and outlier
- OR:
-
Odds ratio
- SNP:
-
Single nucleotide polymorphisms
References
Wongtrakul W, Charoenngam N, Ungprasert P. Increased prevalence of irritable bowel syndrome in migraine patients: A systematic review and meta-analysis. Eur J Gastroenterol Hepatol. 2022;34(1):56–63.
Stern EK, Brenner DM. Gut microbiota-based therapies for irritable bowel syndrome. Clin Transl Gastroenterol. 2018;9(2): e134.
Camilleri M. Irritable Bowel Syndrome: Straightening the road from the Rome criteria. Neurogastroenterol Motil. 2020;32(11): e13957.
Black CJ, Ford AC. Global burden of irritable bowel syndrome: trends, predictions and risk factors. Nat Rev Gastroenterol Hepatol. 2020;17(8):473–86.
Lee C, Doo E, Choi JM. Jang S-h, Ryu H-S, Lee JY, Oh JH, Park JH, Kim YS: The increased level of depression and anxiety in irritable bowel syndrome patients compared with healthy controls: systematic review and meta-analysis. Journal of neurogastroenterology and motility. 2017;23(3):349.
Bonfiglio F, Henström M, Nag A, Hadizadeh F, Zheng T, Cenit M, Tigchelaar E, Williams F, Reznichenko A, Ek WE. A GWAS meta-analysis from 5 population-based cohorts implicates ion channel genes in the pathogenesis of irritable bowel syndrome. Neurogastroenterol Motil. 2018;30(9): e13358.
Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, Enders FT, Ek WE, Schmidt PT, Dlugosz A: Loss-of-function of the voltage-gated sodium channel NaV1. 5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology. 2014;146(7):1659–1668.
Henström M, Diekmann L, Bonfiglio F, Hadizadeh F, Kuech E-M, von Köckritz-Blickwede M, Thingholm LB, Zheng T, Assadi G, Dierks C. Functional variants in the sucrase–isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;67(2):263–70.
Pittayanon R, Lau JT, Yuan Y, Leontiadis GI, Tse F, Surette M, Moayyedi P. Gut microbiota in patients with irritable bowel syndrome—a systematic review. Gastroenterology. 2019;157(1):97–108.
Valeur J, Småstuen MC, Knudsen T, Lied GA, Røseth AG. Exploring gut microbiota composition as an indicator of clinical response to dietary FODMAP restriction in patients with irritable bowel syndrome. Dig Dis Sci. 2018;63(2):429–36.
Simpson CA, Mu A, Haslam N, Schwartz OS, Simmons JG. Feeling down? A systematic review of the gut microbiota in anxiety/depression and irritable bowel syndrome. J Affect Disord. 2020;266:429–46.
Fodor AA, Pimentel M, Chey WD, Lembo A, Golden PL, Israel RJ, Carroll IM. Rifaximin is associated with modest, transient decreases in multiple taxa in the gut microbiota of patients with diarrhoea-predominant irritable bowel syndrome. Gut microbes. 2019;10(1):22–33.
Halkjær SI, Christensen AH, Lo BZS, Browne PD, Günther S, Hansen LH, Petersen AM. Faecal microbiota transplantation alters gut microbiota in patients with irritable bowel syndrome: results from a randomised, double-blind placebo-controlled study. Gut. 2018;67(12):2107–15.
Duan R, Zhu S, Wang B, Duan L. Alterations of gut microbiota in patients with irritable bowel syndrome based on 16S rRNA-targeted sequencing: a systematic review. Clin Transl Gastroenterol. 2019;10(2):e00012–e12.
Lee K, Lim C-Y. Mendelian randomization analysis in observational epidemiology. Journal of Lipid and Atherosclerosis. 2019;8(2):67–77.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–6.
Xu Q, Ni J-J, Han B-X, Yan S-S, Wei X-T, Feng G-J, Zhang H, Zhang L, Li B, Pei Y-F. Causal Relationship Between Gut Microbiota and Autoimmune Diseases: A Two-Sample Mendelian Randomization Study. Front Immunol. 2021;12:5819.
Lee YH. Causal association of gut microbiome on the risk of rheumatoid arthritis: a Mendelian randomisation study. Ann Rheum Dis. 2022;81(1):e3–e3.
Xiang K, Wang P, Xu Z, Hu Y-Q, He Y-S, Chen Y, Feng Y-T, Yin K-J, Huang J-X, Wang J. Causal Effects of Gut Microbiome on Systemic Lupus Erythematosus: A Two-Sample Mendelian Randomization Study. Front Immunol. 2021;12:667097.
Eijsbouts C, Zheng T, Kennedy NA, Bonfiglio F, Anderson CA, Moutsianas L, Holliday J, Shi J, Shringarpure S, Voda A-I. Genome-wide analysis of 53,400 people with irritable bowel syndrome highlights shared genetic pathways with mood and anxiety disorders. Nat Genet. 2021;53(11):1543–52.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, Le Roy CI, Raygoza Garay JA, Finnicum CT, Liu X. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, Smith GD, Sterne JA. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012;21(3):223–42.
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics. 2019;35(22):4851–3.
Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46(6):1734–9.
Larsson SC, Traylor M, Malik R, Dichgans M, Burgess S, Markus HS. Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ. 2017;359:j5375.
Larsson SC, Burgess S. Appraising the causal role of smoking in multiple diseases: A systematic review and meta-analysis of Mendelian randomization studies. EBioMedicine. 2022;82: 104154.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Greco MFD, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–40.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Binda C, Lopetuso LR, Rizzatti G, Gibiino G, Cennamo V, Gasbarrini A. Actinobacteria: a relevant minority for the maintenance of gut homeostasis. Dig Liver Dis. 2018;50(5):421–8.
Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol. 2010;59(9):1114–22.
Wen C, Zheng Z, Shao T, Liu L, Xie Z, Le Chatelier E, He Z, Zhong W, Fan Y, Zhang L. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 2017;18(1):1–13.
Sroka-Oleksiak A, Młodzińska A, Bulanda M, Salamon D, Major P, Stanek M, Gosiewski T. Metagenomic analysis of duodenal microbiota reveals a potential biomarker of dysbiosis in the course of obesity and type 2 diabetes: a pilot study. J Clin Med. 2020;9(2):369.
Krogius-Kurikka L, Lyra A, Malinen E, Aarnikunnas J, Tuimala J, Paulin L, Mäkivuokko H, Kajander K, Palva A. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol. 2009;9(1):1–11.
Wilson B, Rossi M, Kanno T, Parkes GC, Anderson S, Mason AJ, Irving PM, Lomer MC, Whelan K: β-galactooligosaccharide in conjunction with low FODMAP diet improves irritable bowel syndrome symptoms but reduces fecal Bifidobacteria. Official J Am Coll Gastroenterol. 2020;115(6):906–915.
Mazzawi T, Lied GA, Sangnes DA, El-Salhy M, Hov JR, Gilja OH, Hatlebakk JG, Hausken T. The kinetics of gut microbial community composition in patients with irritable bowel syndrome following fecal microbiota transplantation. PLoS ONE. 2018;13(11): e0194904.
Xu X, Dong Q, Zhong Q, Xiu W, Chen Q, Wang J, Zhou Z. The Flavonoid Kurarinone Regulates Macrophage Functions via Aryl Hydrocarbon Receptor and Alleviates Intestinal Inflammation in Irritable Bowel Syndrome. J Inflamm Res. 2021;14:4347.
Hollister EB, Oezguen N, Chumpitazi BP, Luna RA, Weidler EM, Rubio-Gonzales M, Dahdouli M, Cope JL, Mistretta T-A, Raza S. Leveraging human microbiome features to diagnose and stratify children with irritable bowel syndrome. J Mol Diagn. 2019;21(3):449–61.
Pike BL, Paden KA, Alcala AN, Jaep KM, Gormley RP, Maue AC, Christmann BS, Elson CO, Riddle MS, Porter CK. Immunological biomarkers in postinfectious irritable bowel syndrome. J Travel Med. 2015;22(4):242–50.
Liu Y, Zhang L, Wang X, Wang Z, Zhang J, Jiang R, Wang X, Wang K, Liu Z, Xia Z: Similar fecal microbiota signatures in patients with diarrhea-predominant irritable bowel syndrome and patients with depression. Clin Gastroenterol Hepatol. 2016; 14(11):1602–1611. e1605.
Laatikainen R, Jalanka J, Loponen J, Hongisto S-M, Hillilä M, Koskenpato J, Korpela R, Salonen A. Randomised clinical trial: effect of low-FODMAP rye bread versus regular rye bread on the intestinal microbiota of irritable bowel syndrome patients: association with individual symptom variation. BMC Nutrition. 2019;5(1):1–11.
Zhou Y, Zhang F, Mao L, Feng T, Wang K, Wang X, Xu M, Lv B. Bifico Relieves Irritable Bowel Syndrome By Regulating Gut Microbiota Dysbiosis and Inflammatory Cytokines in an Animal Model. Eur J Nutr. 2023;62(1):139-55.
Andriulli A, Bevilacqua A, Palmieri O, Latiano A, Fontana R, Gioffreda D, Castellana S, Mazza T, Panza A, Menzaghi C. Healthy and pro-inflammatory gut ecology plays a crucial role in the digestion and tolerance of a novel Gluten Friendly™ bread in celiac subjects: a randomized, double blind, placebo control in vivo study. Food Funct. 2022;13(3):1299-315.
Wu X, Xu N, Ye Z, Zhao Q, Liu J, Li J, Wu M, Zheng Y, Li X, Li W. Polysaccharide from Scutellaria barbata D. Don attenuates inflammatory response and microbial dysbiosis in ulcerative colitis mice. Int J Biol Macromolecules. 2022;206:1-9.
Chassard C, Dapoigny M, Scott KP, Crouzet L, Del’Homme C, Marquet P, Martin JC, Pickering G, Ardid D, Eschalier A. Functional dysbiosis within the gut microbiota of patients with constipated-irritable bowel syndrome. Aliment Pharmacol Ther. 2012;35(7):828–38.
Rubinstein MR, Baik JE, Lagana SM, Han RP, Raab WJ, Sahoo D, Dalerba P, Wang TC, Han YW. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019;20(4): e47638.
He J, Chu Y, Li J, Meng Q, Liu Y, Jin J, Wang Y, Wang J, Huang B, Shi L: Intestinal butyrate-metabolizing species contribute to autoantibody production and bone erosion in rheumatoid arthritis. Sci Adv. 2022;8(6):eabm1511.
Acknowledgements
We thank the UK Biobank and the Bellygenes initiative for developing and curating their data resources of IBS and we also thank MiBioGen consortium for providing gut microbiota GWAS summary statistics data for our analyses.
Funding
This work was jointly supported by the National Natural Science Foundation of China (82174208, 82074217 and 81973663) and the Research Project of Zhejiang Chinese Medical University (2021JKZKTS001A and 2021JKZKTS004A).
Author information
Authors and Affiliations
Contributions
Bin Liu, Yingying Mao and Guifeng Hao designed the research. Bin Liu, Xiaohui Sun, Hong Yang, Jie Song and Yingying Mao collected and analyzed the data. Bin Liu, Ding Ye, Yingying Mao and Zhixing He performed the literature search. Bin Liu and Guifeng Hao drafted the article. Guifeng Hao and Yingying Mao supervised the study. All authors were involved in writing the paper. All authors contributed to the article and approved the submitted version. Data described in the manuscript, code book, and analytic code will be made available upon request pending.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Effect estimates of the associations between 196 bacterial traits and the risk of IBS in MR analyses among European populations. Table S2. Details of the number of genetic instruments and F-statistic for each cytokine and growth factor.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, B., Ye, D., Yang, H. et al. Assessing the relationship between gut microbiota and irritable bowel syndrome: a two-sample Mendelian randomization analysis. BMC Gastroenterol 23, 150 (2023). https://doi.org/10.1186/s12876-023-02791-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-023-02791-7