
Abstract
Background
Carcinomas of the small bowel are rare tumors usually with dismal prognosis. Most recently, some potentially treatable molecular alterations were described. We emphasize the growing evidence of individualized treatment options in small bowel carcinoma.
Methods
We performed a DNA- based multi-gene panel using ultra-deep sequencing analysis (including 14 genes with up to 452 amplicons in total; KRAS, NRAS, HRAS, BRAF, DDR2, ERBB2, KEAP1, NFE2L2, PIK3CA, PTEN, RHOA, BRCA1, BRCA2 and TP53) as well as an RNA-based gene fusion panel including ALK, BRAF, FGFR1, FGFR2, FGFR3, MET, NRG1, NTRK1, NTRK2, NTRK3, RET and ROS1 on eleven formalin fixed and paraffin embedded small bowel carcinomas. Additionally, mismatch-repair-deficiency was analyzed by checking the microsatellite status using the five different mononucleotide markers BAT25, BAT26, NR-21, NR-22 and NR-27 and loss of mismatch repair proteins using four different markers (MLH1, MSH6, MSH2, PMS2).
Results
In five out of eleven small bowel carcinomas we found potentially treatable genetic alterations. Three patients demonstrated pathogenic (class 5) BRCA1 or BRCA2 mutations – one germline-related in a mixed neuroendocrine-non neuroendocrine neoplasm (MiNEN). Two additional patients revealed an activating ERBB2 mutation or PIK3CA mutation.
Furthermore two tumors were highly microsatellite-instable (MSI-high), in one case associated to Lynch-syndrome. We did not find any gene fusions.
Conclusion
Our results underscore, in particular, the relevance of potentially treatable molecular alterations (like ERBB2, BRCA and MSI) in small bowel carcinomas. Further studies are needed to proof the efficacy of these targeted therapies in small bowel carcinomas.
Similar content being viewed by others


Background
Small intestinal carcinomas account for about 3% of all gastro-intestinal tract tumors [1]. Recent epidemiological data were reported from a Netherland registry showing a 0.7 / 100,000 incidence [2]. Small bowel carcinoma has an annual incidence of about 6 cases per million and is therefore much rarer than colo-rectal carcinomas, which have an incidence of about 420 cases per million inhabitants in the same period (1995–2002) [3,4,5].
The most common tumor location is the duodenum, followed by jejunum and ileum. According to a French study men and women are equally effected. The mean age at occurrence is in their middle to late sixties [6].
The main cause of small bowel carcinoma is unknown. Predisposing factors can be chronic inflammatory bowel syndromes, celiac disease or Lynch-syndrome [3]. In general, the prognosis is worse than for colon carcinoma [7].
According to one publication distal tumor location (ileum) is one predictor for poor survival [8]. On the other hand a recent large study reported that duodenum localisation is a negative predictor of survival after resection of SBA [9].
Thus, there is a strong need for a more effective and personalized systemic treatment option in small bowel carcinoma due to limited effects of standard chemotherapy-based treatments in a metastatic setting.
Seven studies in the past (in years: 1997–2014 analyzed between 15 and 89 tumors each) found 5–35% MSI-small bowel carcinomas. More recently Schrock, A et al. [10] described molecular alterations in 317 small bowel carcinomas as well as Hänninen et al. [1] in additional 160 SBACs. Microsatellite-instability (MSI) was found in 7.6% [10] and 14.2% [1], respectively.
The main molecular mutations considering both publications include (up to): TP53 (48.0%), KRAS (53.6%), APC (26.8%), CDKN2A (14.5%), SMAD4 (17.4%), SOX9 (12.0%) and BRAF (9.1, 10% of these BRAF mutated cases in the study of Schrock showed the common p.V600E mutation whereas Hänninen did not find any BRAF V600E mutations) as well as ERBB2 mutations in 8.2%. [1, 10].
Furthermore, Hänninen et al. described novel candidate driver genes like ACVR1B, BRCA2, and SMARCA4. Copy number gains were observed mainly in KRAS (18.9%), BRAF (17.9%) and PIK3CA (15.3%). Nearly 10% of the Finnish patient cohort suffered from celiac disease. This group revealed a higher amount of MSI tumors [1].
In our study eleven small bowel carcinomas were analyzed focusing on individualized treatment options and on DNA-repair deficiency including BRCA mutations.
Methods
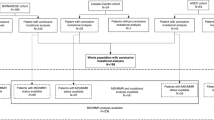
Eleven small bowel carcinomas were selected from the registry of the Institute of Pathology of the University Hospital Cologne, Germany. We identified these cases over a time-frame of six years. We considered primary small bowel tumors (no metastasis) with existing paraffin material for further molecular analyses. Ten of these patients had adenocarcinomas, one a mixed neuroendocrine-non neuroendocrine neoplasm (MiNEN) of the small bowel. All samples were routinely formalin-fixed and paraffin embedded (FFPE) according to local practice.
Parallel sequencing
All tumors were analyzed for a panel of 14 different genes including RAS (K, N, H-RAS), DDR2, BRAF, ERBB2, KEAP1, PIK3CA, NFE2L2, PTEN, TP53, RHOA, BRCA1 and BRCA2 resulting in a total of 452 amplicons. The gene panel includes also 14 different microsatellite regions [11, 12].
Areas of carcinoma were marked on H&E-stained slides by an experienced pathologist and DNA was extracted by manual macro-dissection – details are summarized in [24].
Classification of BRCA variants
According to the established IARC classification each BRCA variant was classified [13] including class 1 variants (not pathogenic) via class 2, class 3, class 4 to class 5 variants (definitively pathogenic). For assessment of variants, the following databases were used:
ARUP BRCA mutation database: http://arup.utah.edu/database/BRCA/
ClinVar database: http://www.ncbi.nlm.nih.gov/clinvar/
Universal mutation database BRCA share: http://www.umd.be/
Leiden Open Variation Database: http://chromium.lovd.nl/LOVD2/
Variants that are not listed in the above mentioned databases were classified according to the ENIGMA criteria (https://enigmaconsortium.org/).
RNA-based fusion panel analysis
Six sections of 10 μm thickness were deparaffinized and the tumor areas were macrodissected from unstained slides using a marked hematoxylin-eosin (H&E) stained slide as a reference. Total nucleic acid was extracted with the Maxwell RSC RNA FFPE Kit on the Maxwell RSC (Promega) according to manufacturer’s instruction, only the DNase solution during the digestion step was replaced by 50 μl water.
Total nucleic acid extracts were quantified with the Qubit RNA BR Assay Kit (Thermo Fisher Scientific) on the Qubit 2.0 Fluorometer (Thermo Fisher Scientific). For the detection of gene fusions the Archer FusionPlex CTL panel (Archerdx, Boulder, CO, USA) was used according to manufacturer’s instructions. In brief, 35–200 ng tNA were target-enriched and prepared cDNA libraries were sequenced on a MiSeq (Illumina). For data analysis and fusion detection the Archer Analysis Software (Archerdx) was used. Strong and weak evidence fusions were evaluated whilst taking into account the read statistics and assay targets.
Immunohistochemical analysis of mismatch-repair deficiency (MMR)
All tumors were stained for MLH1, MSH2, MSH6, PMS2 (using MLH1 (Clone:M1 Ventana), MSH6 (Clone44, Ventana), PMS2 (Clone:EPR3947, Cell Marque), MSH2 (Clone:G219–1129, Cell Marque)) on Ventana Benchmark stainers. 3,3′-Diaminobenzidine (DAB) was used as a chromogen and hematoxylin as a counterstain.
Results
We analyzed formalin-fixed and paraffin-embedded tumor material of eleven patients in total (for patients’ characteristics see Table 1). In about half of our cohort we found potentially treatable genomic alterations in the genes BRCA, ERBB2 and PIK3CA as well as microsatellite instability (see Table 2).
Molecular alterations
Parallel sequencing
Mutational analysis by parallel sequencing was feasible in all eleven tumors. In ten out of eleven tumors the microsatellite status could be determined using five different mononucleotide markers: BAT25, BAT26, NR-21, NR-22 and NR-27 [14].
Four out of eleven tumors revealed a BRCA mutation (two cases with BRCA1 mutations, one case with a BRCA2 mutation and an additional case with known Lynch-syndrome showing co-occurrence of BRCA1 and BRCA2 mutations). According to different databases (UMD, ARUP, ClinVar) all BRCA mutations were classified as pathogenic (class 5) except the Lynch-syndrome associated BRCA-mutations which were classified as class 3 non-pathogenic BRCA mutations. One patient (patient 3) harbors a germline-related BRCA1 mutation and developed a mixed neuroendocrine-non neuroendocrine neoplasm (MiNEN) in his ileum. The patient was microsatellite stable (MSS) and showed a mutation in TP53 (p.C275Y) leading to a non-functional protein. The mutational hot spots of all other analyzed genes in this case were wild type.
The other BRCA mutated carcinomas also presented with co-occuring mutations:
Patient 1 showed a KEAP1 mutation in exon 4: c.1469A > G p.Y490C (allele frequency of 38.6%), a TP53 mutation in exon 8: c.817C > T p.R273C (allele frequency of 36.5%) and wild type sequences in the mutational hot-spots of all other genes tested. For the tumor of this patient we confirmed microsatellite instability (MSI) using the five different markers as describe above and confirmed the loss of DNA repair proteins using immunohistochemistry.
Patient 4 showed a KEAP1 mutation in exon 3: c.959_960GG > TT p.R320L (allele frequency of 29.5%) and wild type sequences in the mutational hot-spots of all other genes tested. This tumor was microsatellite stable.
Altogether, 6/11 tumors (55%) showed a TP53 mutation, 5/11 tumors (45%) showed a KRAS mutation, 4/11 tumors (36%) a BRCA mutation, 3/11 tumors (27%) a PIK3CA mutation, 3/11 tumors (27%) a KEAP1 mutation, 2/10 tumors (20% were MSI and 1/11 carcinomas (9%) a mutation in ERBB2. (Table 2).
RNA-based fusion panel analysis
Due to limited availability of tissue, only six out of eleven tumors were analyzable by RNA sequencing. In none of these tumors a gene fusion was detected with the Archer FusionPlex CTL panel.
Discussion
In this study we were able to confirm the results of Hänninen et al., who described for the first time pathogenic and therapeutically relevant BRCA2 mutations in their analyses of 106 SBAC. Additionally, we found two patients with pathogenic BRCA1 mutations, and one of them turned out to be germline related (patient 3). In patient 4 the BRCA1 mutation was a point mutation with a low allele frequency of 5.5% leading to a truncated protein (p.E1540*) and described as pathogenic in the ARUP and ClinVar databases. In a third patient a somatic truncating BRCA2 mutation in exon 11 with an allele frequency of 38.0% was detected (patient 1, p.N986Ifs*5). According the ENIGMA criteria this truncating mutation is likely pathogenic (class 4). The tumor of a fourth patient with known Lynch-syndrome revealed a BRCA1 as well as a BRCA2 mutation both classified as class 3 mutations and therefore probably not therapeutically important. Both mutations are presumably due to the microsatellite-instability-related higher mutational burden.
DNA repair is essential to maintain DNA integrity – BRCA1 as well as BRCA2 deficient cells show a high degree of chromosomal instability, increasing the risk of malignant transformation [15,16,17,18]. Ovarian carcinomas with a somatic BRCA mutation are likely to respond equally well to therapies that include PARP inhibitors as those with germline related BRCA-mutations [9, 16, 19,20,21,22,23].
In the recent study by Hänninen et al. BRCA mutation were detected for the first time in 7% of 106 patients [1] In our study BRCA mutations were detected with an even higher percentage of 36% (Table 3). However, results could be hampered by the small sample size.
We also confirmed the importance of pathogenic BRCA mutations. In the case of the germline-related BRCA1 mutated MiNEN we could successfully proof the efficacy of a combination of platin-based chemotherapy and the PARP inhibitor olaparib; more than two years after his initial diagnosis of a diffuse metastasized MiNEN (cerebral and different liver metastases) the patient is in a general good condition still without metastases [24].
In addition, first indications are reported that BRCA-mutated ovarian cancers respond well to immuno-checkpoint inhibitors. This is probably due to the higher mutation burden of these tumors compared to BRCA non-mutated ovarian carcinoma [25]. It remains to be shown whether BRCA mutated small bowel adenocarcinoma also benefit from immune-checkpoint inhibition as a second option after PARP inhibition.
ERBB2 is a well-known tyrosine kinase and belongs to the ERBB-family (ERBB1–4). ERBB2 amplification is especially important in breast- and gastric carcinomas and is therapeutically targetable using e.g. the tyrosine-kinase inhibitor trastuzumab. Few publications describe the importance of ERBB2 mutations in small bowel carcinomas [1, 10, 26]. Recent studies by Schrock et al. and Hänninen et al. found comparable results to our study with 8.2 and14% of activating ERBB2 mutations in their patient population [1, 10] In our study we detected in 9% of patient samples an ERBB2 mutation (Table 3). The majority of the ERBB2 mutations clustered into four known hotspots (L755S, was found exclusively in MSI tumors), S310F/Y, R678Q, and V842I). Concurrent hotspot mutations were reported. In concordance with the results mentioned above we could also detect potentially treatment sensitive ERBB2 mutations in our cohort (9%). Our ERBB2 mutated tumor revealed a activating exon 21 mutation (c.2584 A > G p.T862A) which is sensitive to inhibition by neratinib and lapatinib [26].
According to previous studies microsatellite instability (MSI) occurs in a significant number of cases (5–35%). MSI can be germline-related (Lynch-syndrome like in one patient in our cohort) or more frequently somatically induced by an epigenetic silencing of the MLH1 promotor.
Currently, the largest studies by Schrock et al. and Hänninen et al. found high-levels of MSI in SBCAs in 7.6 and 14.1%. We demonstrated a high-level MSI in two out of ten analyzable patients (20%) in our cohort (Table 3). There is growing evidence that microsatellite-instable tumors as well as tumors with a high tumor mutational burden respond well to checkpoint inhibitors and that microsatellite as well as tumor mutational burden status can predict therapy outcome [27].
Results from the keynote studies 158 and 164 confirmed the anti-tumor efficacy of the PD-1-inhibitor pembrolizumab in patients with microsatellite instable colon cancers. Pembrolizumab was approved by the FDA in 2017 for MSI high or mismatch-repair-deficient solid tumors irrespective of tumor origin [28].
The most often altered cancer pathway in SBAC is the PI3K/AKT-pathway. In line with this activating PIK3CA mutations were described in 16% of SBCA by Schrock eal. and were confirmed by us (27%). Tumors driven by an activated PI3K/AKT-pathway might benefit from treatment with a PIK3CA or MEK inhibitor. Currently, clinical trials are ongoing with targeted therapies for PIK3CA mutated tumors of different entities (https://clinicaltrials.gov, for example: NCT02389842, NCT02644122). These therapies are based on convincing preclinical and clinical studies [29, 30].
Beyond the mutations described above (ERBB2, BRCA and PIK3CA) the tumors we investigated revealed additional mutations including in the genes KEAP1, KRAS and TP53 (compare Table 3). There is a growing evidence that TP53 mutated tumors harbor a higher mutational burden and a higher chromosomal instability in comparison to TP53 wild type tumors. Until now nothing is known about different treatment responses to e.g. checkpoint inhibition considering the TP53 mutational status of the tumors. TP53 mutations were detected in our cohort in 55% of samples. This is also in concordance with Schrock et al. and Hänninen et al. Here, PIK3CA mutations were detected with 51 and 48% [1, 10] (Table 3).
Limitations of our study include the small number of cases analyzed. Nevertheless, we were able to demonstrate important and rarely described molecular alterations in SBAC (e.g. BRCA mutations) and could confirm findings of much larger studies (e.g. Schrock et al. and Hänninen et al.) in our small collective. We and other detected potentially targetable molecular alterations with similar percentages (e.g. microsatellite-instability or activating ERBB2-mutations) (Table 3) in SBAC. However, the percentage of BRCA mutations in our smaller sample cohort was higher than previously published. In the future, further clinical validations are needed regarding treatment response. Currently, treatment response against specific genomic alterations in SBACs was extrapolited from other tumor entities like colon or gastric carcinoma.
Parallel sequencing on RNA using the Archer FusionPlex CTL panel did not reveal any gene fusions.
Conclusion
Our results underscore, in particular, the relevance of potentially treatable molecular alterations (like ERBB2, BRCA and MSI) in small bowel carcinomas. Further clinical studies are needed to proof the efficacy of these targeted therapies in small bowel carcinomas.
Abbreviations
- BRCA1:
-
Breast cancer gene 1
- FFPE:
-
Formalin-fixed and paraffin embedded
- H&E:
-
Hematoxylin-eosin
- HR:
-
Homologous recombination
- MiNEN:
-
Mixed neuroendocrine-non neuroendocrine neoplasm
- MMR:
-
Mismatch repair deficiency
- MSI:
-
Microsatellite-instable
- MSS:
-
Microsatellite stable
- PARP:
-
Poly (ADP-Ribose)-Polymerase
- SBAC:
-
Small-bowel adenocarcinoma
- SBCA:
-
Small-bowel carcinoma
References
Hanninen UA, Katainen R, Tanskanen T, Plaketti RM, Laine R, Hamberg J, Ristimaki A, Pukkala E, Taipale M, Mecklin JP, et al. Exome-wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet. 2018;14(3):e1007200.
Legue LM, Bernards N, Gerritse SL, van Oudheusden TR, de Hingh IH, Creemers GM, Ten Tije AJ, Lemmens VE. Trends in incidence, treatment and survival of small bowel adenocarcinomas between 1999 and 2013: a population-based study in the Netherlands. Acta oncologica (Stockholm, Sweden). 2016;55(9–10):1183–9.
Zaaimi Y, Aparicio T, Laurent-Puig P, Taieb J, Zaanan A. Advanced small bowel adenocarcinoma: molecular characteristics and therapeutic perspectives. Clin Res Hepatol Gastroenterol. 2016;40(2):154–60.
Faivre J, Trama A, De Angelis R, Elferink M, Siesling S, Audisio R, Bosset JF, Cervantes A, Lepage C, Group RW. Incidence, prevalence and survival of patients with rare epithelial digestive cancers diagnosed in Europe in 1995-2002. Eur J Cancer. 2012;48(10):1417–24.
Center MM. Jemal a, smith RA, Ward E: worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59(6):366–78.
Lepage C, Bouvier AM, Manfredi S, Dancourt V, Faivre J. Incidence and management of primary malignant small bowel cancers: a well-defined French population study. Am J Gastroenterol. 2006;101(12):2826–32.
Overman MJ, Hu CY, Kopetz S, Abbruzzese JL, Wolff RA, Chang GJ. A population-based comparison of adenocarcinoma of the large and small intestine: insights into a rare disease. Ann Surg Oncol. 2012;19(5):1439–45.
Aparicio T, Svrcek M, Zaanan A, Beohou E, Laforest A, Afchain P, Mitry E, Taieb J, Di Fiore F, Gornet JM, et al. Small bowel adenocarcinoma phenotyping, a clinicobiological prognostic study. Br J Cancer. 2013;109(12):3057–66.
Wilhelm A, Galata C, Beutner U, Schmied BM, Warschkow R, Steffen T, Brunner W, Post S, Marti L. Duodenal localization is a negative predictor of survival after small bowel adenocarcinoma resection: a population-based, propensity score-matched analysis. J Surg Oncol. 2018;117(3):397–408.
Schrock AB, Devoe CE, McWilliams R, Sun J, Aparicio T, Stephens PJ, Ross JS, Wilson R, Miller VA, Ali SM, et al. Genomic profiling of small-bowel adenocarcinoma. JAMA Oncol. 2017;3(11):1546–53.
Kloth M, Ruesseler V, Engel C, Koenig K, Peifer M, Mariotti E, Kuenstlinger H, Florin A, Rommerscheidt-Fuss U, Koitzsch U, et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan-HER inhibitors in lynch and lynch-like colorectal cancer. Gut. 2016;65(8):1296–305.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44(10):1104–10.
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282–91.
Nardon E, Glavac D, Benhattar J, Groenen PJ, Hofler G, Hofler H, Jung A, Keller G, Kirchner T, Lessi F, et al. A multicenter study to validate the reproducibility of MSI testing with a panel of 5 quasimonomorphic mononucleotide repeats. Diagn Mol Pathol. 2010;19(4):236–42.
Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta. 2014;1846(1):201–15.
Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449–55.
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74.
Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96(1):11–5.
O'Sullivan CC, Moon DH, Kohn EC, Lee JM. Beyond breast and ovarian cancers: PARP inhibitors for BRCA mutation-associated and BRCA-like solid tumors. Front Oncol. 2014;4:42.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd, Abkevich V, Potter J, Pruss D, Glenn P, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly(ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–6.
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–75.
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–90.
Quaas A, Waldschmidt D, Alakus H, Zander T, Heydt C, Goeser T, Daheim M, Kasper P, Plum P, Bruns C, et al. Therapy susceptible germline-related BRCA 1-mutation in a case of metastasized mixed adeno-neuroendocrine carcinoma (MANEC) of the small bowel. BMC Gastroenterol. 2018;18(1):75.
Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, Adams SF. CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian Cancer. Cancer Immunol Res. 2015;3(11):1257–68.
Laforest A, Aparicio T, Zaanan A, Silva FP, Didelot A, Desbeaux A, Le Corre D, Benhaim L, Pallier K, Aust D, et al. ERBB2 gene as a potential therapeutic target in small bowel adenocarcinoma. Eur J Cancer. 2014;50(10):1740–6.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–51.
Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev. 2014;40(8):980–9.
Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141(2):160–71.
Planck M1, Ericson K, Piotrowska Z, Halvarsson B, Rambech E, Nilbert M. Microsatellite instability and expression of MLH1 and MSH2 in carcinomas of the small intestine. Cancer. 2003;97(6):1551–7.
Acknowledgements
None.
Funding
None.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
AQ, CH, SM, DW, HA, TZ made substantial contributions to conception and design. CB, TG, WR, ABr, MS, PK, NH were responsible for acquisition of data. RB, AQ, CH, ABu were responsible for analyses and interpretation of data. All authors have been involved in drafting the manuscript or revising it critically for important intellectual content All authors given final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Procedures were followed as outlined in accordance with ethical standards formulated in the Helsinki Declaration 1975 (and revised in 1983). Patients gave their written consent to usage of their tumor specimens, an approval was obtained from the University of Cologne Ethics Committee (reference number: 13–091).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Quaas, A., Heydt, C., Waldschmidt, D. et al. Alterations in ERBB2 and BRCA and microsatellite instability as new personalized treatment options in small bowel carcinoma. BMC Gastroenterol 19, 21 (2019). https://doi.org/10.1186/s12876-019-0942-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-019-0942-z