
Abstract
Background
Although deleterious mutations in interleukin-10 and its receptor molecules cause severe infantile-onset inflammatory bowel disease, there are no reports of mutations affecting this signaling pathway in Japanese patients. Here we report a novel exonic mutation in the IL10RA gene that caused unique splicing aberrations in a Japanese patient with infantile-onset of inflammatory bowel disease in association with immune thrombocytopenic purpura and a transient clinical syndrome mimicking juvenile myelomonocytic leukemia.
Case presentation
A Japanese boy, who was the first child of non-consanguineous healthy parents, developed bloody diarrhea, perianal fistula, and folliculitis in early infancy and was diagnosed with inflammatory bowel disease. He also developed immune thrombocytopenic purpura and transient features mimicking juvenile myelomonocytic leukemia. The patient failed to respond to various treatments, including elemental diet, salazosulfapyridine, metronidazole, corticosteroid, infliximab, and adalimumab. We identified a novel mutation (c.537G > A, p.T179T) in exon 4 of the IL10RA gene causing unique splicing aberrations and resulting in lack of signaling through the interleukin-10 receptor. At 21 months of age, the patient underwent allogeneic hematopoietic stem cell transplantation and achieved clinical remission.
Conclusions
We describe a novel exonic mutation in the IL10RA gene resulting in infantile-onset inflammatory bowel disease. This mutation might also be involved in his early-onset hematologic disorders. Physicians should be familiar with the clinical phenotype of IL-10 signaling defects in order to enable prompt diagnosis at an early age and referral for allogeneic hematopoietic stem cell transplantation.
Similar content being viewed by others


Background
Interleukin-10 (IL-10), an anti-inflammatory cytokine, binds to 2 receptors, namely 2 alpha molecules (IL-10RA/IL-10R1) and 2 beta molecules (IL-10RB/IL-10R2) [1, 2]. IL-10 signaling plays a key role in maintaining immune homeostasis in the gastrointestinal tract. Accordingly, defects of IL10, IL10RA, or IL10RB genes cause very early-onset inflammatory bowel disease (IBD) including infantile-onset IBD (IOIBD) [3–5]. Patients with mutations in IL10 or IL10 receptor (IL10R) genes present with severe colitis, perianal disease and folliculitis manifesting in the first months of life. These patients are refractory to immunosuppressive therapies such as corticosteroids, methotrexate, thalidomide, and anti-tumor necrosis factor-alpha (TNF-α) antibodies, yet, allogeneic hematopoietic stem cell transplantation (HSCT) has been shown to be curative in these conditions [3–5]. To date over 40 patients of various ethnicities with IL10/IL10R deficiency have been reported [3–14]. However, reports on IL-10 signaling pathway defects in the Japanese population are lacking. Here we describe a Japanese IOIBD patient with a novel exonic mutation in the IL10RA gene causing unique splicing aberrations. Interestingly, this patient also developed immune thrombocytopenic purpura (ITP) and transient abnormalities mimicking juvenile myelomonocytic leukemia (JMML) in early infancy.
Case presentation
A Japanese boy was born by spontaneous vaginal delivery at 41 weeks of gestation, with a birth weight of 2834 g. He was the first child of non-consanguineous healthy parents. At 2 months of age, he presented with hepatosplenomegaly, purpura, leukocytosis (white blood cell count, 2.5 × 1010 /L), monocytosis (monocytes, 2.0 × 109 /L), anemia (hemoglobin, 87 g/L), and thrombocytopenia (platelets, 3.6 × 1010 /L). Bone marrow examination disclosed hypercellularity without excessive blasts or abnormal appearance of megakaryocytes. Cytogenetic analysis indicated a normal male karyotype, and colony assay of bone marrow mononuclear cells showed spontaneous colony formation and high sensitivity to granulocyte-macrophage colony stimulating factor. Genetic analysis revealed no reported mutations in PTPN11, NRAS, KRAS, or CBL genes. While his clinical manifestations met diagnostic criteria for JMML [15], his symptoms and blood test results normalized spontaneously.
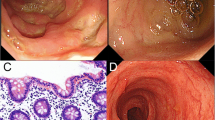
At 6 months of age he developed bloody diarrhea, perianal fistula, and folliculitis. Endoscopy showed erosive changes, multiple aphthous ulcers, and longitudinal ulcerations of the colon. Elevated C-reactive protein and increased erythrocyte sedimentation rate suggested the presence of a chronic inflammatory disorder. Immune work-up was normal, including T and B lymphocytes numbers, proliferative response of peripheral blood mononuclear cells (PBMCs) to phytohemagglutinin, serum immunoglobulin concentrations, neutrophil phagocytic capacity and microbicidal activity. The Wiskott-Aldrich syndrome protein was expressed normally in PBMCs.
Based on clinical, laboratory, and endoscopic findings, the patient was diagnosed with Crohn’s disease, which was treated with an elemental diet, salazosulfapyridine, metronidazole, and a corticosteroid. At the same time, he again developed thrombocytopenia (platelets, 3.2 × 1010 /L) and was diagnosed with ITP since megakaryocytes in the bone marrow were normal and platelet-associated immunoglobulin G was elevated (90 ng/107 cells) [16]. Treatment with high-dose intravenous immunoglobulin G led to prompt normalization of platelets count, but did not lead to any improvement in bloody diarrhea, fistula, or folliculitis. The patient also had recurrent fevers and was treated with systemic antibiotics.
Given lack of colitis improvement, at 9 months of age, he began treatment with infliximab (IFX), achieving partial response of bloody diarrhea, perianal fistula, and folliculitis. At the age of 14 months, adalimumab (ADA) was substituted for IFX because of an allergic reaction to infusions. From that point he was treated with a maintenance regimen of ADA, an elemental diet, and salazosulfapyridine. Growth and development then entered the normal range, although bloody diarrhea and folliculitis persisted. Given his clinical features, an IL-10 signaling defect was suspected and targeted sequencing confirmed an IL10RA mutation.
At 21 months of age, the patient underwent allogeneic HSCT using umbilical cord blood from a matched unrelated donor at Kyushu University Hospital, achieving rapid and sustained complete chimerism and clinical remission. Currently, at the age of 24 months, the patient is in sustained clinical remission since 2 months after HSCT.
Identification of novel IL10RA mutation and IL-10 receptor defect
To detect mutations of IL10RA and IL10RB, we performed direct sequence analysis using genomic DNA isolated from PBMCs from the patient and his parents (see Additional files 1 and 2). We identified a novel homozygous and heterozygous mutation (c.537G > A, p.T179T) at the 3' end of exon 4 of IL10RA in the patient and his parents, respectively (Fig. 1a). Because the nucleotides at the end of the exon are involved in the splicing process, we examined whether the c.537G > A mutation generated splicing variants by reverse-transcription polymerase chain reaction (RT-PCR) analysis using 3 primer sets: set A, to amplify exons 2 and 3 as a control; set B, to detect deletion or insertion near the boundary between exons 4 and 5; and set C, to amplify exons 3 and 4 (upper panel in Fig. 1b). We detected an additional small PCR product in the patient, as well as the product of expected size using set B (compare band #3 with #2, lower panel in Fig. 1b). Moreover, we found a much smaller product in the patient using set C (band #4, lower panel in Fig. 1b). These results suggested that the exonic c.537G > A mutation generated at least 2 abnormal transcripts of the IL10RA gene. We further investigated the nucleotide sequence of these PCR products. As shown in Fig. 1c, product #2, which showed expected size, possessed a c.537G > A mutation but indicated intact splicing between exon 4 and 5. However, product #3 showed an 18-base deletion at the 3' end of exon 4, which was generated by activation of a nearby cryptic splice donor site. This 18-base deletion was predicted to cause loss of 6 amino acids (174-VPGNFT-179) from the IL-10RA molecule. Moreover, product #4 showed complete deletion of exon 4 (170 bases). This exon 4 skipping caused a frameshift, with translational arrest at a site 7 amino acids distant. Thus, the exonic c.537G > A mutation induced aberrant splicing in the IL10RA gene, which would cause loss of function.

Identification of the IL10RA mutation associated with infantile-onset inflammatory bowel disease (IOIBD). a A c.537G > A mutation at the 3' end of exon 4 of the IL10RA gene in the family of our patient with IOIBD. Arrows indicate site of mutation. Parts of sequences of exon 4 and intron 4 are shown in upper case and lower case, respectively. b RT-PCR analysis for the IL10RA gene in the patient with c.537G > A mutation. Schematic representation of positions of the primers for RT-PCR and the c.537G > A mutation is given in the upper panel. RT-PCR products from the c.537G > A mutation of the IL10RA gene are shown in the lower panel. The expected 216-bp and 495-bp products of the IL10RA exons 4-5 and exons 2-5, respectively, were detected in a healthy control and the patient, while additional shorter products were observed in the patient (bands 3 and 4). c Sequence analysis of RT-PCR products indicated that the c.537G > A mutation caused 2 kinds of splicing variants; an 18 bp deletion of the 3' end of exon 4 and skipping of exon 4 (170 bp)
To determine whether the novel mutation identified in the patient resulted in defective signaling function involving the IL-10R, PBMCs obtained from the patient were compared with those from his father, who served as a healthy control. Cells were stimulated with IL-10 as previously described [13] (see Additional file 3). IL-10 stimulation of the patient’s PBMCs failed to induce phosphorylation of signal transducer and activator of transcription 3 (pSTAT3), which is a key transcription factor downstream of the IL-10R. IL-6-induced pSTAT3, which served as an internal positive control, was intact (right panel in Fig. 2). In contrast, both IL-10 stimulation and IL-6 stimulation of the control cells led to pSTAT3 formation (left panel in Fig. 2). These results suggest that the mutation resulted in loss of normal function of the IL-10R.

Loss of function of the IL-10 receptor. Functional analysis of IL-10 receptor complex was performed by determining signal transducer and activator of transcription 3 (STAT3) phosphorylation using flow cytometry. Peripheral blood mononuclear cells from the patient (right panel) and his father (left panel) were stimulated for 15 minutes with IL-10 (20 ng/mL) or IL-6 (20 ng/mL) or kept unstimulated, and later fixed, permeabilized and stained for phosphorylated STAT3 (pSTAT3), which is downstream of the IL-10 and IL-6 receptor complexes
Discussion
Loss of function of IL-10 and IL-10R molecules causes refractory colitis with folliculitis and perianal disease, manifesting very early in infancy [3–5]. Here we report a Japanese IOIBD patient with a novel exonic mutation (c.537G > A, p.T179T) in exon 4 of the IL10RA gene that produced uniquely abnormal transcripts. Also in early infancy, the patient developed ITP and a transient clinical syndrome with features mimicking JMML.
From early infancy, the patient manifested refractory colitis, perianal disease and folliculitis, in addition to associated hematologic disorders, which may also be the result of the IL10RA genetic defect. Expression of IL-10 in PBMCs has been reported to correlate to some extent with disease activity in children with chronic active ITP [17]. IL-10 normally exerts negative feedback control that limits activation of Th1 cells as well as monocytes and macrophages [18]. In vitro, IL-10 has been found to inhibit cell growth and granulocyte/macrophage colony stimulating factor production in chronic myelomonocytic leukemia cells, as well as inhibiting cytokine production and growth in JMML cells [19, 20]. Neven et al and Shouval et al reported that patients with IL-10R deficiency tend to develop non-Hodgkin B-cell lymphomas [13, 21]. These findings suggest that IL-10 signaling defects might lead not only to development of lymphoma but also to ITP and JMML-like states. However, there is a possibility that there may be another gene involved here as the remainder of this patient’s genes have not been tested.
We identified a point mutation, c.537G > A, in the exon adjacent to a splice donor site whose consensus sequence is MAG|GURAGU (M represents A or C, while R represents a purine), extending from position -3 to position +6 relative to the exon–intron junction. Within the consensus sequence, mutations affecting GU residues at positions +1 and +2 are most common followed by mutations at position +5 [22]. In our patient, a relatively rare -1G > -1A homozygous mutation resulted in aberrant splicing manifest as disease. The AG|GURAGU motif is recognized by the U1 small nuclear ribonucleoprotein (snRNP) to define the exon-intron boundary in the first step of splicing process [23]. When the motif is changed, U1 snRNP would not recognize the exon-intron junction properly and would bind to other regions with similar sequence. In the exon 4 of IL10RA, there is AGGTGCCA sequence at 18 bases upstream of exon 4-intron 4 boundary where c.537G > A is located. This sequence might be recognized by U1 snRNP, and the splicing variant of 18-base deletion at the 3' end of exon 4 was generated in our patient. However, the AGGTGCCA sequence in exon 4 is not completely matched to the conserved AGGURAGU motif, so that another splicing variant, exon 4 skipping, might occur alternatively. This type of splicing aberration is not specific to the case in this study because we obtained similar results in the patients affected with familial hemophagocytic lymphohistiocytosis previously [24, 25].
According to the database which predicts splice-altering single nucleotide variants (dbscSNV: https://www.solvebio.com/library/dbscSNV), the IL10RA c.537G > A substitution shows high probability of affecting splicing (the Ada score = 0.99996). This in silico analysis strongly supports our results that the c.537G > A mutation generated splicing variants. Although the c.537G > A nucleotide change is not found in the three databases (1000 Genome: http://www.1000genomes.org/, ExAC: http://exac.broadinstitute.org/, or NHLBI Exome Variant Server: http://evs.gs.washington.edu/EVS/), the frequency of this variant is shown to be 0.0019 in the Human Genetic Variation Database which contains genetic variations determined by exome sequencing of 1208 Japanese (http://www.genome.med.kyoto-u.ac.jp/SnpDB/). This suggests that the c.537G > A mutation would be detected in other Japanese IOIBD patients in future.
The c.537G > A mutation caused an in-frame deletion of 18 bp of exon 4 in the IL10RA gene that leads to loss of 6 amino acids (174-VPGNFT-179). Structural analysis of the IL-10/IL-10RA/IL-10RB complex suggests that these amino acids compose a hydrophobic area that could importantly influence crucial interactions with IL-10RB molecules [26]. Because sequential assembly of IL10-RA and IL-10RB is required for IL-10 cellular response [27], disruption of stable interactions between these molecules could lead to a loss of IL-10 signaling, resulting in IOIBD in our patient. Although we also identified the normal splicing PCR product in our patient, we speculated that the normal splicing produces a small amount of IL-10RA and cannot rescue the IL-10 signaling in his macrophages and T cells.
Conclusions
We identified a novel exonic mutation (c.537G > A, p.T179T) in the IL10RA gene that caused unique splicing aberrations likely to lead to IOIBD. IL-10R genetic defects also might be involved in hematologic disorders during infancy such as ITP and JMML-like disorders. Physicians should be familiar with the clinical phenotype of IL-10 signaling defects in order to enable prompt diagnosis at an early age and referral for allogeneic HSCT, before later complication occur, such as development of lymphoma.
Consent
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal. This study was approved by the Institutional Review Board at Kurume University, Japan and the Boston Children’s Hospital, MA.
Abbreviations
- IL-10:
-
interleukin-10
- IL-10RA:
-
IL-10 receptor alpha subunit
- IL-10RB:
-
IL-10 receptor beta subunit
- IBD:
-
inflammatory bowel disease
- IL-10R:
-
interleukin-10 receptor
- IOIBD:
-
infantile-onset inflammatory bowel disease
- TNF-α:
-
tumor necrosis factor-alpha
- HSCT:
-
hematopoietic stem cell transplantation
- ITP:
-
immune thrombocytopenic purpura
- JMML:
-
juvenile myelomonocytic leukemia
- PBMCs:
-
peripheral blood mononuclear cells
- IFX:
-
infliximab
- ADA:
-
adalimumab
- RT-PCR:
-
reverse-transcription polymerase chain reaction
- pSTAT3:
-
phosphorylation of signal transducer and activator of transcription 3
- snRNP:
-
small nuclear ribonucleoprotein
References
Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. 2014;122:177–210.
Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–19.
Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45.
Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–55.
Engelhardt KR, Shah N, Faizura-Yeop I, Kocacik Uygun DF, Frede N, Muise AM, et al. Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J Allergy Clin immunol. 2013;131:825–30.
Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Infant colitis—it’s in the genes. Lancet. 2010;376:1272.
Begue B, Verdier J, Rieux-Laucat F, Goulet O, Morali A, Canioni D, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106:1544–55.
Moran CJ, Walters TD, Guo CH, Kugathasan S, Klein C, Turner D, et al. IL-10R polymorphisms are associated with very-early onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–23.
Pigneur B, Escher J, Elawad M, Lima R, Buderus S, Kierkus J, et al. Phenotypic characterization of very early-onset IBD due to mutations in the IL10, IL10 receptor alpha or beta gene: a survey of the GENIUS Working Group. Inflamm Bowel Dis. 2013;19:2820–8.
Murugan D, Albert MH, Langemeier J, Bohne J, Puchalka J, Järvinen PM, et al. Very early onset inflammatory bowel disease associated with aberrant trafficking of IL-10R1 and cure by T cell replete haploidentical bone marrow transplantation. J Clin Immunol. 2014;34:331–9.
Shim JO, Seo JK. Very early-onset inflammatory bowel disease (IBD) in infancy is a different disease entity from adult-onset IBD; one form of interleukin-10 receptor mutations. J Hum Genet. 2014;59:337–41.
Lee CH, Hsu P, Nanan B, Nanan R, Wong M, Gaskin KJ, et al. Novel de novo mutations of the interleukin-10 receptor gene lead to infantile onset inflammatory bowel disease. J Crohns Colitis. 2014;8:1551–6.
Shouval DS, Ebens CL, Murchie R, McCann K, Rabah R, Klein C, et al. Large B-cell lymphoma in an adolescent patient with IL-10 receptor deficiency and history of infantile inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2014. [Epub ahead of print]
Beser OF, Conde CD, Serwas NK, Cokugras FC, Kutlu T, Boztug K, et al. Clinical features of interleukin 10 receptor gene mutations in children with very early-onset inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2015;60:332–8.
Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125:1083–90.
Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–93.
Mouzaki A, Theodoropoulou M, Gianakopoulos I, Vlaha V, Kyrtsonis MC, Maniatis A. Expression patterns of Th1 and Th2 cytokine genes in childhood idiopathic thrombocytopenic purpura (ITP) at presentation and their modulation by intravenous immunoglobulin G (IVIg) treatment: their role in prognosis. Blood. 2002;100:1774–9.
Elenkov IJ, Chrousos GP. Stress hormones, Th1/ Th2 patterns, pro/anti-inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metab. 1999;10:359–68.
Geissler K, Ohler L, Födinger M, Virgolini I, Leimer M, Kabrna E, et al. Interleukin 10 inhibits growth and granulocyte/macrophage colony-stimulating factor production in chronic myelomonocytic leukemia cells. J Exp Med. 1996;184:1377–84.
Iversen PO, Hart PH, Bonder CS, Lopez AF. Interleukin (IL)-10, but not IL-4 or IL-13, inhibits cytokine production and growth in juvenile myelomonocytic leukemia cells. Cancer Res. 1997;57:476–80.
Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood. 2013;122:3713–22.
Buratti E, Chivers M, Královicová J, Romano M, Baralle M, Krainer AR, et al. Aberrant 5' splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35:4250–63.
Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. 2004;5:389–96.
Yamamoto K, Ishii E, Sako M, Ohga S, Furuno K, Suzuki N, et al. Identification of novel MUNC13-4 mutations in familial haemophagocytic lymphohistiocytosis and functional analysis of MUNC13-4-deficient cytotoxic T lymphocytes. J Med Genet. 2004;41:763–7.
Nagai K, Yamamoto K, Fujiwara H, An J, Ochi T, Suemori K, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PLoS One. 2010;5:e14173.
Pletnev S, Magracheva E, Wlodawer A, Zdanov A. A model of the ternary complex of interleukin-10 with its soluble receptors. BMC Struct Biol. 2005;5:10.
Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. 1997;16:5894–903.
Acknowledgements
We thank the patient and his parents for their cooperation, as well as Ms. Miki Sonoda at Kurume University School of Medicine (for technical help of genetic analysis), Dr. Masahiro Migita at the Japanese Red Cross Kumamoto Hospital (for providing early-infancy patient information), and Drs. Hideki Muramatsu and Seiji Kojima at Nagoya University Graduate School of Medicine (for carrying out the colony assay and genetic analysis related to JMML).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TY, TM, MI, HT, SS, and KY contributed to the concept and design of the study. TY, TM, YT, KE, KM, MI, HT, and YY contributed to treatment of the patient. TY, TM, YT, KE, DS, AG, SS, and KY contributed to analysis and interpretation of the data. TY, TM, KM, MI, HT, DS, SS, YY, and KY contributed to writing the manuscript. All authors contributed to the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Genetic analysis of the IL10RA and IL10RB genes. (DOCX 13 kb)
Additional file 2:
Primer sets for mutation screening of IL10RA and IL10RB genes. (DOCX 15 kb)
Additional file 3:
Functional assay of the IL-10 receptor. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yanagi, T., Mizuochi, T., Takaki, Y. et al. Novel exonic mutation inducing aberrant splicing in the IL10RA gene and resulting in infantile-onset inflammatory bowel disease: a case report. BMC Gastroenterol 16, 10 (2016). https://doi.org/10.1186/s12876-016-0424-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-016-0424-5