
Abstract
Background
Aquilegia is a model system for studying the evolution of adaptive radiation. However, very few studies have been conducted on the Aquilegia mitochondrial genome. Since mitochondria play a key role in plant adaptation to abiotic stress, analyzing the mitochondrial genome may provide a new perspective for understanding adaptive evolution.
Results
The Aquilegia amurensis mitochondrial genome was characterized by a circular chromosome and two linear chromosomes, with a total length of 538,736 bp; the genes included 33 protein-coding genes, 24 transfer RNA (tRNA) genes and 3 ribosomal RNA (rRNA) genes. We subsequently conducted a phylogenetic analysis based on single nucleotide polymorphisms (SNPs) in the mitochondrial genomes of 18 Aquilegia species, which were roughly divided into two clades: the European-Asian clade and the North American clade. Moreover, the genes mttB and rpl5 were shown to be positively selected in European-Asian species, and they may help European and Asian species adapt to environmental changes.
Conclusions
In this study, we assembled and annotated the first mitochondrial genome of the adaptive evolution model plant Aquilegia. The subsequent analysis provided us with a basis for further molecular studies on Aquilegia mitochondrial genomes and valuable information on adaptive evolution in Aquilegia.
Similar content being viewed by others

Background
Mitochondria are semiautonomous organelle that carry out respiratory metabolism and provides energy for life activities [1]. Compared with those in the chloroplast genome, in plant mitochondrial genomes, genome rearrangements, repeat sequence recombination, and gene integration/loss/transfer/duplication occur frequently, greatly affecting the normal function of mitochondrial genes [2]. The mitochondrial genome structure is often described as circular, but its real structure appears to be diverse, such as circular, linear, and complex branched [3]. Additionally, the sequence evolution of the plant mitochondrial genome has been very slow, and the nucleotide synonym substitution rate has ranged from several to tens of times lower than that of plant chloroplast and nuclear genomes [4]. With the rapid development of genome sequencing technologies and assembly methods, the whole mitochondrial genomes of many plants have been assembled [5, 6]. The mitochondrial genomes play a key role in the adaptive evolution of plants, especially in response to abiotic stresses. They are involved in energy production, metabolism, regulation of PCD, and ROS production [7]. For example, the mitochondrial genomes of high-altitude plants may have experienced adaptative shrinkage to better survive extreme environments on the plateau [5]; similarly, the mitochondrial genome of Geoffroea decorticans (Gillies ex Hook. & Arn.) Burkart plants likely underwent structural changes facilitating their adaptation to the extreme conditions in the Atacama Desert [8].
Aquilegia L. (columbine) is a perennial herb of the Ranunculaceae family [9]. Along with several new species being reported, approximately 110 species of columbine taxa have been found, which is an ideal material for studying adaptive radiation evolution [10]. The chromosomal-level genome and chloroplast genome of Aquilegia have previously been reported, but the phylogenetic relationships of Aquilegia based on different genomes are controversial, which have made the evolution of adaptive radiation in Aquilegia unclear. However, systematic research and evolutionary analysis of the Aquilegia mitochondrial genome are lacking [11, 12]. The assembly and analysis of the mitochondrial genome could provide a new perspective and reference for the identification of the phylogeny of Aquilegia. This approach can help us to realize and understand how the mitochondrial genome changed during the adaptive evolution of Aquilegia.
In this study, we assembled the complete mitochondrial genome of Aquilegia amurensis Kom. and analyzed codon usage bias to reveal characteristics of mitochondrial genome. In order to understand the phylogenetic relationships and adaptive evolution A. amurensis of Aquilegia from the mitochondrial genome perspective. First, we identified SNPs from 18 species including North American, European, and Asian species. Then, phylogenetic trees were constructed based on the SNPs to lay a foundation for inferring the evolutionary history of Aquilegia. Finally, we explored positive selection in Aquilegia to advance the current genetic and evolutionary understanding of this genus.
Materials and methods
Plant material and mitochondrial genome assembly
Fresh leaves of A. amurensis were collected from Changbai Mountain (N 42.053, E 128.048; Jilin Province, China). The identification of plants was conducted by Hongxing Xiao. The deposition number was 220730-0101 and the voucher number was NENU403102, these materials were deposited in the Northeast Normal University Herbarium in Changchun, China. High-quality DNA was extracted using the modified cetyltrimethylammonium bromide (CTAB) method [13], and evaluated using a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and a NanoDrop One spectrophotometer (Thermo Fisher Scientific). PacBio HiFi sequencing was performed on the PacBio Sequel II platform (Pacific Biosciences, CA, USA) according to the manufacturer’s instructions.
The de novo assembly of mitochondrial genome was performed using Flye [14] based on the long subreads. In terms of parameter setting, the assembly was set four times in the minimum overlapping sequences of 1000, 3000, 5000 and 10,000, and the other parameters were set to their defaults. We used Bandage [15] to visualize the mitochondrial genome sketch. The mitochondrial sequences of A. amurensis were selected with BLASTn [16] using the complete mitochondrial sequences of Liriodendron tulipifera L. (NC_021152.1) as query. Due to the complexity of the sketch and the existence of some repeated sequences, BWA [17] was used to map the HiFi data to the graphical mitochondrial genome fragments, which excluded the repeated sequences to ensure that the assembly result was supported by a larger number of reads.
The mitochondrial genome of A. amurensis was initially annotated using GeSeq tools [18] on the MPI-MP CHLOROBX website (https://chlorobox.mpimp-golm.mpg.de) with the reference mitogenome (L. tulipifera, GenBank: NC_021152.1). The tRNA genes were identified using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE) [19], while the rRNA genes were annotated using BLASTn software. Apollo [20], an interactive tool that allows biological experts to improve these approximations by viewing and independently evaluating the data supporting each annotation, was used to manually correct annotation errors in each mitochondrial genome. The mitochondrial map was drawn using Organellar Genome DRAW (OGDRAW) [21].
Codon usage bias and repeated sequence analysis
Phylosuite v1.1.16 [22] was used to extract the protein-coding sequences of the A. amurensis mitochondrial genome. MEGA v7.0.26 [23] was used to analyze basic sequence information such as base composition, nucleotide sequence information sites, start codons, and stop codons and to calculate relative synonymous codon usage (RSCU). The results were visualized using the R package ggplot2 [24].
Simple sequence repeat (SSRs), tandem repeats and dispersed repeats were detected using the MISA web server (https://webblast.ipk-gatersleben.de/misa/) [25], the Tandem Repeats Finder web server v4.09 (https://Tandem.bu.edu/trf/trf.unix.help.html/) [26] and the REPuter Network web server (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) [27], respectively. The results were visualized using the R package ggplot2 [24]. In addition, we used ROUSFinder.py to analyze non-tandem repeats [28].
Phylogenetic analysis
To investigate the phylogenetic relationships of Aquilegia species based on the mitochondrial genome, we collected whole-genome sequencing data from 18 Aquilegia species, including A. sibirica Lam., A. ecalcarata Maxim., and A. amurensis. A. parviflora Ledeb., A. rockii Munz, A. viridiflora Pall., A. yabeana Kitag., A. oxysepala var. kansuensis Brühl, A. oxysepala var. oxysepala Trautv. et Mey., A. japonica Nakai & Hara, A. canadensis L., A. coerulea E. James, A. barnebyi Munz, A. longissimi A. Gray, A. chrysantha A. Gray, A. formosa Fisch. ex DC, A. aurea Janka, A. vulgaris Richardson, from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/sra) (Table S1).
The raw data were processed in two steps: adapter sequences in the reads were trimmed and then reads that contained more than 50% low quality bases (quality value ≤ 5) were removed. The remaining sequencing reads from Aquilegia species were aligned separately to the mitochondrial genome of A. amurensis using BWA [17]. The readgroups were added using the AddOrReplaceReadGroups module in GATK v.4.1.8.0 [29], and the MarkDuplicates module was subseqently used to mark the duplicates. The index was subsequently built through SAMtools v.0.1.18 [30]. The SelectVariants module in GATK v.4.1.8.0 was used to extract the original SNPs. SNPs with mapping quality ≥ 40 and a SNP quality ≥ 30 were retained. VCFtools v0.1.13 [31] was used to remove SNPs with a minor allele frequency (MAF) of 0.05 or less, more than 2 alleles, a mean sequencing depth of less than 50, or missing rate of more than 0.5. Moreover, only homozygous SNPs were retained [32]. Semiaquilegia adoxoides Makino (SRR437677) and Paraquilegia microphylla Drumm. et Hutch (SRR26400723, unpublished) were considered outgroups [11, 33] (Table S1). The maximum likelihood (ML) method was used for phylogenetic analysis using IQTREE v2.2.2.6 with 1000 ultrafastbootstrap replicates [34]. According to the Bayesian information criterion (BIC), the best-fit model for the SNP datasets was the TVM + F + G4 model. Meanwhile, Bayesian inference (BI) method was used to reconstruct a phylogenetic tree using MrBayes v3.2 [35] under the GTR + G + I model. The Markov Chain Monte Carlo (MCMC) analyses were run for 1000,00 generations. The trees were sampled every 100 generations, and the first 25% of the trees were discarded as burn-in. The final trees were visualized using Figtree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and iTOL v4.0 (https://itol.embl.de/itol.cgi). The phylogenetic trees of the different genome were compared using the R package phytools [36].
Selection pressure analysis
Using the annotated information of A. amurensis as a reference and SNPs, we obtained the aligned CDS of the mitochondrial genomes. The nonsynonymous substitution rate (dN), synonymous substitution rate (dS), and dN/dS ratio of each protein-coding gene were calculated by the yn00 module using the Codeml program in the PAML package [37]. The dN/dS ratio is an important index of selection pressure. A dN/dS ratio > 1, dN/dS ratio = 1, and dN/dS ratio < 1 indicated positive, neutral, and negative selection, respectively [38]. Moreover, the selection pressure of European species and Asian species, the selection pressure of North American species, and the selection pressure between the above two groups were compared. The results were visualized using the R package ggplot2 [24].
Results
Complete mitochondrial genome sequence of A. amurensis
A total of 22,851,573 subreads were obtained from the PacBio Sequel II platform and the N50 of the subreads was 20,484 bp. Based on long-read data from the PacBio Sequel platform, the main structure of the mitochondrial genome was multibranch structure. We obtained six nodes of the A. amurensis mitochondrial genome, which were named on each node and formed overlapping areas with each other along the connecting lines to form a complex genome structure (Fig. S1). Even contig4-contig5-contig6 and contig1-contig2-contig4 can form a closed ring structure, where contig2 and contig6 were not included. The above three different types of assembly results were all confirmed computationally (Fig. S1). To succinctly describe the mitochondrial genome and avoid redundancy of partial sequences, we used a circular ring structure (contig1-contig3-contig5-contig4) and two linear fragments (contig2 and contig6) as representative sequences of the mitochondrial genome.
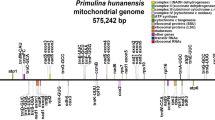
The total length of mitochondrial genome of A. amurensis was 538,736 bp, and the GC content was 46.21%. Chromosome 1 had a circular structure, a length of 462,264 bp, and a GC content of 46.22%. Chromosome 2 and 3 were linear fragments. The lengths of these fragments were 50,804 bp and 25,668 bp, respectively, and the GC content was 44.92% and 48.64%, respectively (Fig. 1). Moreover, a total of 24 tRNA genes, 3 rRNA genes and 33 unique protein-coding genes were annotated, including 24 unique mitochondrial core genes and 9 noncore genes (Table S2). There were five ATP synthase genes (atp1, atp4, atp6, atp8 and atp9); nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9); four cytochrome C biogenetic genes (ccmB, ccmC, ccmFC and ccmFN); three cytochrome C oxidase genes (cox1, cox2 and cox3), one membrane transport-protein-gene (mttB); one mature enzyme-encoding gene (matR); and one panthenol-cytochrome C reductase gene (cob) annotated in the mitochondrial genome. The noncore genes included three ribosomal large subunit genes (rpl5, rpl10, and rpl16), six ribosomal small subunit genes (rps4, rps7, rps10, rps12, rps13, and rps14) (Fig. 1, Table S2).

Schematic of the mitochondrial genome of A. amurensis. Genes belonging to different functional groups are color-coded
Relative synonymous codon usage of A. amurensis mitochondrial genome
Codon usage bias is thought to be the result of a relative balance within the cell over a long period of evolutionary selection. An RSCU value greater than 1 was considered to indicate the beneficial effect of amino acids. There were 27 codons for which RSCU > 1 (Fig. 2a). The RSCU values of the start codon AUG and tryptophan (UGG) were both 1. The remaining mitochondrial protein-coding genes exhibited general codon usage (Fig. 2a). The stop codon had a high preference for UAA, and its RSCU value was 1.8, which was the highest among the mitochondrial protein-coding genes. Alanine (Ala) had the second highest preference for GCU codons, with the an RSCU value of 1.61(Fig. 2a). Notably, cysteine (Cys) and phenylalanine (Phe) did not have strong codon usage with maximum RSCU values less than 1.2 (Fig. 2a). The results showed that A or T nucleotides were used more frequently at the third codon position than were C or G nucleotides.

Relative synonymous codon usage (a), SSRs (b) and other repeats (c) in the mitochondrial genome of A. amurensis
Repeat sequence analysis of the A. amurensis mitochondrial genome
In total, we detected 156 SSRs, including 141 SSRs on chromosome 1, 10 SSRs on chromosome 2 and 5 SSRs on chromosome 3. In addition to chromosome 3, monomeric and dimeric nucleotide repeats on chromosomes 1 and 2, which contained 54 (38.30%) and 5 (50.00%), respectively (Table S3). There were 39 tandem repeats in the mitochondrial genome with more than 73% match and a length between 12 and 72 bp (Table S4). For nontandem repeats, 718 pairs of other repeats with a length greater than or equal to 30 bp were found, including 365 pairs of palindromic repeats, 352 pairs of forward repeats, and 1 pair of complementary repeats; moreover, no reverse repeats or long repeats (> 1000 bp) were found.
On chromosome 1, adenine (A) monomeric repeats accounted for 62.96% (17) of the twenty-seven monomeric SSRs (Fig. 2b). TA and CT repeats were the most common types of dimeric SSRs, accounting for 51.85% of the dimeric SSRs (Table S3). On chromosome 1, there were two hexametric SSRs. Thirty-three tandem repeats were matching degree greater than 72% and length ranging from 13 to 72 bp (Table S4). A total of 708 dispersed repeats with lengths greater than or equal to 30 were observed, including 364 palindromic repeats, 343 forward repeats and 1 complementary repeat, and no reverse repeats were detected (Fig. 2c). The longest palindromic repeat was 310 bp and the longest forward repeat was 231 bp (Table S5). On chromosome 2 and 3, there were few SSRs, tandem repeats or dispersed repeats. Moreover, reverse repeats or complementary repeats were not found.
Phylogenetic analysis of Aquilegia
To explore the phylogenetic relationships between Aquilegia species and to clarify the phylogenetic position of A. amurensis, trees were constructed using 1533 SNPs of mitochondrial genome and 97 SNPs located in genes. The ML tree and Bayesian tree showed the same topology, and the posterior probabilities of the Bayesian tree for each lineage were greater (Fig. S2). Based on the phylogenetic tree of mitochondrial genome, approximately two clades were identified within Aquilegia. One clade included all the species from Asia and Europe, while the other clade included six species distributed in North America. Despite the low ultrafastbootstrap (ufbs value = 42) and posterior probability (PP value = 78) of the European-Asian clade, there were strong supports for the European-Asian clade in the phylogenetic trees based on chloroplast and nuclear genomes data (Fig. 3). The clade containing European and Asian species was divided into two subclades. A. amurensis and A. parviflora were in the same subclade, while the topology supported A. japonica and A. oxysepala var. oxysepala as sister branches, and A. vulgaris from Europe had a common ancestor. A. ecalcarata from the eastern group and the western group were divided into two subclades. A. ecalcarata from the eastern group was closely related to A.sibirica, and A. ecalcarata from the western group formed a single clade with A. rockii and A. oxysepala var. kansuensis (Fig. S2).

Comparison of phylogenetic relationship in Aquilegia. Comparison of phylogenetic tree between mitochondrial and chloroplast genome (a); comparison of phylogenetic relationship between mitochondrial and nuclear genome (b). The phylogenetic trees of mitochondrial genome, chloroplast genome and nuclear genome were constructed based on 1533, 599 and 363,842 SNPs, respectively. The blue, orange, and green dots represent species from Asia, Europe, and North America, respectively. The gray dots represent outgroup species. The ML ultrafastbootstrap (ufbs) and BI posterior probability (PP) values are indicated above the branches. “*” are ufbs or PP of 100
Selection pressure analysis of Aquilegia mitochondrial protein-coding genes
It is important to determine the nonsynonymous substitution rate (dN) and synonymous substitution rate (dS) as these parameters are highly important for understanding the evolutionary dynamics of protein-coding sequences. The rps14 gene was positively selected, as indicated by a dN/dS ratio greater than 1.0 in all three comparisons. (Table S6). In addition, the genes mttB, rpl5, rps4, and rps13 were positively selected in European and Asian species (Fig. 4). According to the comparison between European-Asian species and North American species, the genes rps4 and rps12 were also under positive selection (Fig. 4). Compared with those of other protein-coding genes, the genes atp9, ccmC, ccmFC, cox1, cox3, nad1, nad3, nad4L, nad6, nad7, rpl16, and rps7 had significantly lower dN/dS ratios, indicating that their functions were highly conserved (Table S6).

dN/dS ratios of protein-coding genes in the mitochondrial genome of Aquilegia. The upper and lower limits and circles in the thick lines represent the upper quartile, lower quartile, and median of the pairwise dN/dS ratios of each gene, respectively
Discussion
Characterization of the A. amurensis mitochondrial genome
The mitochondrion is a semiautonomous organelle with its own genetic material and genetic system, and it has the ability to independently replicate and inherit [1]. The best-known function of mitochondria is the conversion of proton concentration gradients to ATP for biological activity through oxidative phosphorylation. Because of the energy metabolism of mitochondrial DNA, its role in adaptive evolution has attracted much attention. In particular, during evolution, mitochondria play a crucial role in the internal regulation of organisms under harmful environmental conditions such as hypoxia and low temperature [39]. Here, we sequenced, assembled, and analyzed the mitochondrial genome of A. amurensis to understand how Aquilegia adaptively evolved from the perspective of the mitochondrial genome. After the repeat regions were excluded from the HiFi data, we obtained one circular contig and two linear contigs.
Changes in the size and structure of plant mitochondrial genomes are obvious, but functional genes remain conserved [40]. The mitochondrial genome of the common ancestor of angiosperms consists of 41 protein-coding genes [41]. Approximately 33 out of the 41 protein-coding genes were detected in the Aquilegia mitochondrial genome. Compared with those in A. thaliana, the genes rpl2, rps3, and rps7 were missing. The total length of mitochondrial genome of Aquilegia was greater than that of Arabidopsis thaliana L., Oryza sativa L., Triticum aestivum L. and other model plants [42,43,44], but lower than that of Zea mays L [45]. There were fewer genes in A. amurensis than in the above model plants. The transfer of mitochondrial genes to the nuclear genome may explain the above results, meaning that gene transfer between the cell nucleus and cytoplasm may be more frequent in A. amurensis. Gene transfer from mitochondrial to the nuclear genome has been common during plant evolution [46].
Mitochondria and chloroplast are self-renewing organelles with independent genomes. The total length of the mitochondrial genome was 538 kb, which was three times that of the chloroplast genome. However, the gene density of the mitochondrial genome was one-third that of chloroplast genome [12]. The GC content of the chloroplast genome is often lower than that of the mitochondrial genome [6].
The RSCU is closely related to the evolution of organisms. On the one hand, codon preference can affect amino acid sequences, protein structure and function, thus affecting the adaptability and survival ability of organisms. On the other hand, codon preference can also reflect the evolutionary process and trend of the genome. The PCGs of A. amurensis mitochondria typically begin with ATG start codons and preferentially end with A or T in their stop codon, which is similar to the codon preference of angiosperms [47]. The relative usage of the synonymous codons of A. amurensis and O. sativa was similar [48]. The results indicated a strong A/T bias in the third codon position of A. amurensis mitochondrial protein-coding genes, which is commonly observed in plant mitochondrial genomes [49]. SSRs are believed to play a major role in inducing the genetic variation underlying adaptation [50]. A connection between SSRs and stress resistance in plants has been reported. In A. amurensis, the highest proportions of SSRs in the nuclear genome and chloroplast genome were monomer nucleotide repeats and dimer nucleotide repeats [12], while tetrameric nucleotide repeats accounted for the highest proportion of SSRs in the mitochondrial genome. An increase in the number of nucleotide repeats in SSRs in the mitochondrial genome may promote the stress resistance of Aquilegia species during evolution [51].
The phylogeny of Aquilegia based on mitochondrial genomes
In this study, we constructed phylogenetic trees based on SNPs of mitochondrial genomes to provide a new perspective on the phylogeny of Aquilegia. The results showed broadly similar phylogenetic trees based on nuclear or chloroplast genome data. The species of Aquilegia were roughly divided into two clades: the European-Asian clade and the North American clade, but the phylogenetic positions of some species were not completely consistent [11, 12, 52]. For example, phylogenetic trees based on mitochondrial and chloroplast genomes (unpublished) supported that A. parviflora was located in the European–Asian clade, while phylogenetic trees based on the nuclear genome (unpublished) showed that A. parviflora was closely related to North American species (Fig. 3). According to the phylogenetic tree of the mitochondrial genome, A. yabeana and A. parviflora were sister branches, while according to the phylogenetic tree of nuclear and chloroplast genomes (unpublished), A. yabeana was closely related to A. rockii, A. oxysepala var. kansuensis and A. ecalcarata (Fig. 3). Although both the chloroplast and mitochondrial genomes of Aquilegia were maternally inherited, they exhibited different phylogenetic relationships and differed from those of phylogenetic studies based on nuclear genomes. Therefore, the mitochondrial genome of plants could be an ideal molecular marker for phylogenetic studies, and an important tool for exploring biological evolution.
There were some inconsistencies in the phylogenetic trees of the mitochondrial, chloroplast and nuclear genomes [53, 54]. Aquilegia species were mainly cross-pollinated under natural conditions, characterized by extensive hybridization and high genetic diversity [55]. Some inconsistencies between the morphological and molecular phylogenies may indicate that hybridization played a major role in the evolution of the genus [56]. Organelle capture is the process by which the chloroplast genome of a plant infiltrates from one plant species to another after hybridization or backcrossing with the parental population. Organelle capture could occur frequently in species with sympatric distributions or contact zones and reproductive compatibility, which may explain the phylogenetic inconsistencies of Aquilegia. In addition, limited sampling, incomplete lineage classification, and differences in evolutionary rates could explain the phylogenetic reconstruction of the unclear and two organelle genomes [54, 57].
Adaptative evolution of Aquilegia
dN/dS analysis of the mitochondrial genomes of Aquilegia revealed that most of the genes were under negative selection during evolution, indicating that the protein-coding genes of the mitochondrial genome were well-conserved. The observed pattern of dN/dS ratios is concordant with the expectations concerning the mitochondrial genomes for which negative selection was reported to be the predominant force of evolution [58]. This functional constraint of mitochondrial genes is associated with the important role of the organelle which is sustained by purifying selection, and is responsible for maintaining the long-term stability of biological structures by eliminating deleterious variations. As a consequence, protein-coding genes of mitochondrial genomes are conserved in land plants [59]. These protein-coding genes with dN/dS <1 may play important roles in stabilizing the normal function of mitochondria. For example, the protein encoded by the gene atp9 is one of the chains of the nonenzymatic membrane component (F0) of mitochondrial ATPase; the gene nad2 is critical to the mitochondrial NADH dehydrogenase homologous subunits; and the gene ccmC may be involved in the export of heme to the mitochondrion for the biogenesis of c-type cytochromes [60,61,62].
However, the dN/dS ratios of the genes mttB, rpl5, rps4 and rps13 were > 1, according to the selection pressure analysis of European-Asian species, indicating that these protein-coding genes were positively selected during evolution. The genes rps4, rps12 and rps13, like the rps14 gene, which was selected for all three comparisons, are involved in the small subunits of ribosomes. Moreover, they have different effects on the translation of mitochondrial mRNA. The mttB gene encodes a transport membrane protein, that is involved in the molecular functions of proton transmembrane transfer and proton-dynamic dependent protein transmembrane transfer functional protein and encodes a TATC-like protein (also known as orf X) [63]. Although little is known about the actual mitochondrial function of the mttB gene, this gene may be essential for mitochondrial function. The mttB gene was not lost in the mitochondrial genomes of 280 angiosperms species, which were intact [64]. The mttB gene controls the activity of trimethylamine methyltransferase to affect methylation, and is associated with metabolic alterations and oxidative stress [65]. For example, under high-salt conditions, the mttB gene was highly upregulated [66]. Transposable elements (TEs) in the plant mitochondrial genome can help plants adapt to the high-altitude environment, and the number of transposons around the mttB gene was greatest, which also indicated that the mttB gene was related to the adaptive evolution of plants [67]. The protein encoded by the rpl5 gene is part of the 5 S ribonucleoprotein particle (5 S RNP), which is an important component of the ribosomal large subunit (LSU) and is required for rRNA [68]. The rpl5 gene is a ribosomal protein-encoding gene with a high substitution rate and genetic variation in the organelle genome [69]. Previous studies have shown that mutations in large subunits of ribosomal protein genes inhibit sensitivity to temperature [70]. The rpl5 gene was also positively selected during the evolution of Paropyrum anemonoides (Kar. & Kir.) Ulbr, and may be developing novel stress resistance mechanisms in Ranunculaceae plants [71]. The genes mttB and rpl5 are highly sensitive to responses to stress and signaling molecules, indicating that these genes encode proteins that exhibit stress-ameliorating effects in addition to housekeeping [72]. Positive selection of these two genes involves fixing beneficial variation induced by environmental factors in a population and promoting the emergence of new phenotypes that can adapt to particular environmental conditions [73]. Adaptive evolution in European and Asian species may have been driven by a variety of environmental factors. In general, the genes mttB and rpl5 might have developed novel functions related to stress resistance in European species and Asian species under positive selection pressure, enabling the species to be widely distributed [74].
Conclusions
We successfully assembled and annotated the mitochondrial genome of A. amurensis and performed extensive analyses based on annotated genes. We obtained a circular chromosome and two linear chromosomes when repeated sequences were deleted, for a total length of 538,736 bp. We annotated 60 genes, including 33 protein-coding genes, 24 tRNA genes and 3 rRNA genes. Compared with the mitochondrial genome of model plants, the gene density of the mitochondrial genome of A. amurensis was lower. The phylogenetic tree of Aquilegia species based on mitochondrial genome showed similar topological structure to that of the nuclear and chloroplast genomes. The Aquilegia species were divided into two clades: the European-Asian clade and the North American clade, but some species had different phylogenetic positions. Finally, by analyzing selection pressure, we found that the mitochondrial genes mttB and rpl5 may have contributed to the adaptive evolution of European and Asian species against adverse environments. This study complements the genetic knowledge available for the genus Aquilegia and provides new insights into the adaptive evolution of plants.
Data availability
The A. amurensis mitochondrial genome sequence was deposited in the GenBank database (accession number OR818043, OR818044 and OR818045). Accession numbers for reference sequences downloaded from Genbank were provided in Table S2.
Abbreviations
- tRNA:
-
Transfer RNA
- rRNA:
-
Ribosomal RNA
- SNP:
-
Single nucleotide polymorphism
- SSR:
-
Simple sequence repeat
- CTAB:
-
Cetyltrimethylammonium bromide
- RSCU:
-
Relative synonymous codon usage ratios
- d N /d S :
-
Ratios Nonsynonymous to synonymous substitution ratios
References
Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505(7483):335–43.
Jang W, Lee HO, Kim J-U, Lee J-W, Hong C-E, Bang K-H, Chung J-W, Jo I-HJA. Complete mitochondrial genome and a set of 10 novel kompetitive allele-specific PCR markers in ginseng (Panax Ginseng CA Mey.). 2020, 10(12):1868.
Zhang S, Wang J, He W, Kan S, Liao X, Jordan DR, Mace ES, Tao Y, Cruickshank AW, Klein R et al. Variation in mitogenome structural conformation in wild and cultivated lineages of sorghum corresponds with domestication history and plastome evolution. BMC Plant Biol 2023, 23(1).
Varré J-S, d’Agostino N, Touzet P, Gallina S, Tamburino R, Cantarella C, Ubrig E, Cardi T, Drouard L, Gualberto JM. Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int J Mol Sci. 2019;20(19):4788.
Xiong Y, Yu Q, Xiong Y, Zhao J, Lei X, Liu L, Liu W, Peng Y, Zhang J, Li D. The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Frontiers in plant science 2021, 12.
Ma Q, Wang Y, Li S, Wen J, Li Q. Assembly and comparative analysis of the first complete mitochondrial genome of Acer Truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol 2022, 22(1).
Stechmann A, Hamblin K, Perez-Brocal V, Gaston D, Richmond GS, Van der Giezen M, Clark CG, Roger AJ. Organelles in Blastocystis that blur the distinction between mitochondria and hydrogenosomes. Curr Biol. 2008;18(8):580–85.
Contreras-Diaz R, Carevic FS, van den Brink L. Comparative analysis of the complete mitogenome of Geoffroea decorticans: a native tree surviving in the Atacama Desert. Front Genet 2023, 14.
Munz PA. Aquilegia: the cultivated and the wild columbines. Bailey Hortorium; 1946.
Kramer EM, Hodges SA. Aquilegia as a model system for the evolution and ecology of petals. Philosophical Trans Royal Soc B-Biological Sci. 2010;365(1539):477–90.
Fior S, Li M, Oxelman B, Viola R, Hodges SA, Ometto L, Varotto C. Spatiotemporal reconstruction of the Aquilegia rapid radiation through next-generation sequencing of rapidly evolving cpDNA regions. New Phytol. 2013;198(2):579–92.
Zhang W, Wang H, Dong J, Zhang T, Xiao H. Comparative chloroplast genomes and phylogenetic analysis of Aquilegia. Appl Plant Sci 2021, 9(3).
Arseneau J-R, Steeves R, Laflamme M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol Ecol Resour. 2017;17(4):686–93.
Kolmogorov M, Yuan J, Yu L, Pevzner PA. Assembly of long error-prone reads using repeat graphs. Cold Spring Harbor Lab 2018(5).
Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: interactive visualization of de novo genome assemblies. Bioinf 2015, 31(20):3350–52.
Chen Y, Ye W, Zhang Y, Xu Y. High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 2015;43(16):7762–68.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Michael T, Pascal L, Tommaso P, Ulbricht-Jones ES, Axel F, Ralph B, Stephan G. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res 2017(W1):W1.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997(5).
Lewis SE, Searle S, Harris N, Gibson M, Clamp ME. Apollo: a sequence annotation editor. Genome Biol Evol. 2002;3(12):RESEARCH0082.
Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019;47(W1):W59–W64.
Zhang D, Gao F, Jakovli I, Zou H, Wang GT. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 2020;20(1):348–55.
Sudhir K, Glen S, Koichiro T. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biology Evol. 2016;33(7):1870–74.
Ito K, Murphy D. Application of ggplot2 to pharmacometric graphics. CPT:pharmacometrics & systems pharmacology 2013, 2:e79.
Sebastian B, Thomas T, Thomas M, Uwe S, Martin M. MISA-web: a web server for microsatellite prediction. Bioinf 2017, 33(16):2583.
Gary B. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–42.
Wynn EL, Christensen AC. Repeats of unusual size in Plant mitochondrial genomes: identification, incidence and evolution. G3-Genes genomes Genetics 2019, 9(2):549–59.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data P: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–79.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, Depristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Scarcelli N. Population Genomics of Organelle genomes in Crop plants. Population Genomics; 2020.
Zhai W, Duan X, Zhang R, Guo C, Li L, Xu G, Shan H, Kong H, Ren Y. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol Phylogenet Evol. 2019;135:12–21.
Lam-Tung N, Schmidt HA, Arndt VH, Quang MB. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biology Evol. 2015;32(1):268–74.
Huelsenbeck JP. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Revell LJ. Phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ. 2024;12:e16505–e05.
Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol BiolEvolution. 2007;24:1586–91.
Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biology Evol. 2002;16(6):908.
Onukwufor JO, Kibenge F, Stevens D, Kamunde C. Hypoxia-reoxygenation differentially alters the thermal sensitivity of complex I basal and maximal mitochondrial oxidative capacity. Volume 201. Comparative Biochemistry and Physiology a-Molecular & Integrative Physiology; 2016. pp. 87–94.
Hong Z, Liao X, Ye Y, Zhang N, Yang Z, Zhu W, Gao W, Sharbrough J, Tembrock LR, Xu D, et al. A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia Odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics. 2021;22(1):672.
Mower JP, Sloan DB, Alverson AJ. Plant mitochondrial genome diversity: the genomics revolution. 2012.
Unseld M, Marienfeld JR, Brandt P, Brennicke A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat Genet. 1997;15(1):57–61.
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Volume 268. Proteomics: Molecular Genetics Genomics; 2002. pp. 434–45. 4.
Yasunari O, Yukiko Y, Koji M, Akira K, Toru T, Takashi S, Naohiko M, Shuhei N, Chiharu N, Naoki M. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005;33(19):6235–50.
Clifton SW, Minx P, Fauron MR, Gibson M, Allen JO, Sun H, Thompson M, Barbazuk WB, Kanuganti S, Tayloe C. Sequence and comparative analysis of the Maize NB mitochondrial genome. Plant Physiol. 2004;136(3):3486–503.
Adams KL, Qiu YL, Stoutemyer M, Palmer JD. Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci U S A. 2002;99(15):9905–12.
Zhou P, Zhang Q, Li F, Huang J, Zhang M. Assembly and comparative analysis of the complete mitochondrial genome of Ilex metabaptista (Aquifoliaceae), a Chinese endemic species with a narrow distribution. BMC Plant Biol 2023, 23(1).
Jin G, Wang L, Long L, Wu F, Tang Y, Qin J, Wei D, Huang Q, Su W. Analysis of codon usage bias in the mitochondrial protein-coding genes of Oryza rufipogon. Plant Sci J. 2019;37(2):188–97.
Shidhi PR, Biju VC, Anu S, Vipin CL, Deelip KR, Achuthsankar SN. Genome characterization, comparison and phylogenetic analysis of complete mitochondrial genome of evolvulus alsinoides reveals highly rearranged gene order in Solanales. Life-Basel. 2021;11(8):769.
Kashi Y, King DG. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006;22(5):253–59.
Freitas KEJ, Busanello C, Viana VE, Pegoraro C, Victoria FD, Maia LC, Oliveira AC. An empirical analysis of mtSSRs: could microsatellite distribution patterns explain the evolution of mitogenomes in plants? Functional & Integrative Genomics 2022, 22(1):35–53.
Filiault DL, Ballerini ES, Mandakova T, AkoZ G, Derieg NJ, Schmutz J, Jenkins J, Grimwood J, Shu S, Hayes RD et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. Elife 2018, 7.
Li Z, Guo X, Price M, Zhou S, He X. Phylogenetic position of Ligusticopsis (Apiaceae, Apioideae): evidence from molecular data and carpological characters. Aob Plants 2022, 14(2).
Ren T, Li Z, Xie D, Gui L, Peng C, Wen J, He X. Plastomes of eight Ligusticum species: characterization, genome evolution, and phylogenetic relationships. BMC Plant Biol 2020, 20(1).
Hodges SA, Derieg NJ. Adaptive radiations: from field to genomic studies. Proc Natl Acad Sci USA. 2009;106:9947–54.
Garcia-Jacas N, Uysal T, Romashchenko K, Suarez-Santiago VN, Ertugrul K, Susanna A. Centaurea revisited: a molecular survey of the Jacea group. Ann Botany. 2006;98(4):741–53.
Rutherford S, Rossetto M, Bragg JG, McPherson H, Benson D, Bonser SP, Wilson PG. Speciation in the presence of gene flow: population genomics of closely related and diverging Eucalyptus species. Heredity. 2018;121(2):126–41.
Bazin EG, Galtier S. Population size does not influence mitochondrial genetic diversity in animals. Science. 2006;312:570–72.
Myszczynski K, Gorski P, Slipiko M, Sawicki J. Sequencing of organellar genomes of Gymnomitrion concinnatum (Jungermanniales) revealed the first exception in the structure and gene order of evolutionary stable liverworts mitogenomes. BMC Plant Biology 2018, 18(321).
Wang S, Li J, Zhu J. Comparative analysis of differential expression of mitochondrial genes in three plants at low temperature. Guihaia. 2020;40(8):1140–50.
Busi MV, Gómez-Casati DF, Mariano PA, Araya A, Zabaleta E. Nuclear-encoded mitochondrial complex I gene expression is restored to normal levels by inhibition of unedited ATP9 transgene expression in Arabidopsis thaliana. Plant Physiol Biochem. 2006;44(1):1–6.
Stoll K, Jonietz C, Binder S. In Arabidopsis thaliana two co-adapted cyto-nuclear systems correlate with distinct ccmC transcript sizes. Plant J. 2015;81(2):247–57.
Bentolila S, Oh J, Hanson MR, Bukowski R. Comprehensive high-resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genet 2013, 9(6).
Mower JP, Palmer JD. Patterns of partial RNA editing in mitochondrial genes of Beta vulgaris. Mol Genet Genomics. 2006;276(3):285–93.
Ticak T, Kountz DJ, Girosky KE, Krzycki JA, Ferguson DJ Jr. A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc Natl Acad Sci USA. 2014;111(43):E4668–E76.
Liu X, Luo Y, Mohamed OA, Liu D, Wei G. Global transcriptome analysis of Mesorhizobium alhagi CCNWXJ12-2 under salt stress. BMC Microbiol 2014, 14.
Xiong Y, Yu Q, Xiong Y, Zhao J, Lei X, Liu L, Liu W, Peng Y, Zhang J, Li D et al. The complete mitogenome of Elymus sibiricus and insights into itsevolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front Plant Sci 2022, 12.
Zhang H, Liu J, Dang Q, Wang X, Chen J, Lin X, Yang N, Du J, Shi H, Liu Y et al. Ribosomal protein RPL5 regulates colon cancer cell proliferation and migration through MAPK/ERK signaling pathway. BMC Mol Cell Biology 2022, 23(1).
Liu L, Du Y, Folk RA, Wang S, Soltis DE, Shang F, Li P. Plastome evolution in Saxifragaceae and multiple plastid capture events involving Heuchera and Tiarella. Frontiers in Plant Science 2020, 11.
De Zoysa T, Hauke AC, Iyer NR, Marcus E, Ostrowski SM, Fay JC, Phizicky EM. A connection between the ribosome and two S. pombe tRNA modification mutants subject to rapid tRNA decay. bioRxiv: the preprint server for biology 2023.
Yisilam G, Liu Z, Turdi R, Chu Z, Luo W, Tian X. Assembly and comparative analysis of the complete mitochondrial genome of Isopyrum anemonoides (Ranunculaceae). PLoS ONE. 2023;18(10):e0286628–e28.
Moin M, Bakshi A, Saha A, Dutta M, Madhav SM, Kirti PB. Rice ribosomal protein large subunit genes and their spatio-temporal and stress regulation. Front Plant Sci 2016, 7.
Desai MM, Fisher DS. Beneficial mutation-selection balance and the effect of linkage on positive selection. Genet 2007, 176(3):1759–98.
Li J, Xu Y, Shan Y, Pei X, Yong S, Liu C, Yu J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254(2).
Funding
This research was supported by National Natural Science Foundation of China (grant nos. 32070244 and 32300187).
Author information
Authors and Affiliations
Contributions
H.X. and H.W. conceptualized the basic idea and plan the study; H.W. and L.X. collected the materials; L.X, J.W., and T.Z. participated in the data analysis; L.X. and H.W. prepared the manuscript; and all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
This study’s material collections and experimental research were obtained with the permission of Changbai Mountain Administration Commission, in accordance with relevant institutional, national and international guidelines and legislation. And we only collected plant leaves during material collection.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Figure S1.
Mitochondrial genome sketch of A. amurensis (node ID marked in the figure). The orange color represents a major circular genome structure after resolving the duplicate region based on HiFi data
Supplementary Material 2: Figure S2.
Phylogenetic relationships based on the mitochondrial genome of Aquilegia. The ML ultrafastbootstrap (ufbs) and BI posterior probability (PP) values are indicated above the branches. “*” are ufbs or PP of 100
Supplementary Material 3: Table S1.
Information about the Aquilegia sequences data previously published. Table S2. Annotated genes list in the mitochondrial genome of A. amurensis. Table S3. SSRs in the mitochondrial genome of A. amurensis. Table S4. Tandem repeat sequences in the mitochondrial genome of A. amurensis
Supplementary Material 4: Table S5.
Dispersed repeat sequences in the mitochondrial genome of A. amurensis
Supplementary Material 5: Table S6.
dN/dS ratios of each gene in the mitochondrial genome of Aquilegia
Supplementary Material 6: Table S7.
Nontandem repeat sequences in the mitochondrial genome of A. amurensis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, L., Wang, J., Zhang, T. et al. Characterizing complete mitochondrial genome of Aquilegia amurensis and its evolutionary implications. BMC Plant Biol 24, 142 (2024). https://doi.org/10.1186/s12870-024-04844-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-04844-9